|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
![]()
Authors: F. Cazals and G. Carriere and A. Chevallier and T. Dreyfus
The description of molecular systems encompasses several aspects (geometry, topology, biophysics), see SBL: terminology and concepts.
This package solely handles the topology, a.k.a. the molecular covalent structure (MCS).
In short, the template class SBL::IO::T_Molecular_covalent_structure_loader is the orchestra conductor, as it commands the construction of a MCS based on two main ingredients:
This user manual is following is organized as follows:
In all generality, a PDB or mmCIF file contains proteins and nucleic acids, and possibly other molecular species. We note that proteins and nucleic acids are special cases of biopolymers–see package Linear_polymer_representation, namely molecules assembled by gluing units also called residues. We also note that depending on the biophysical application of interest, various pieces of information may be attached to a given atom.
To present the MCS data structure, we distinguish four levels:
More specifically:
Atoms/particles: the type of atom/pseudo atom used, and the associated pieces of information. In fact, in the sequel, Atom is referred to as ParticleInfo.
Molecular covalent structure (MCS): the graph used to represent the topology. This graph is typically built from the sequence of units of the molecule (a.a. for proteins, bases for nucleic acids). Therefore, we provide MCS for boths.
The builder: the algorithm building and filling a specific MCS. Builder are therefore dedicated to proteins or nucleic acids which are linear biopolymers – see package Linear_polymer_representation.
The manipulation of atoms involves the following sets of indices:
PDB serial number. The integer used to index the ATOM in a PDB file. Note that entities with a serial number are: ATOM, HETATM, ANISOU, and TER.
Atom serial number/atomid. The index assigned to the atom generally by the parser loading the PDB/cif file. May not coincide with the PDB serial number, due in particular to serial numbers of TER tags used to separate chains.
Atom linear position: atom position in the structure, once gaps have been removed. A plain linear ordering from 0 to n-1, if there are n atoms. This index is used to access the position i.e. the Cartesian coordinates of the atom in the conformation of the molecule – see below. In using libcifpp, the linear position is the atom serial number minus 1.
Particle representation, aka Vertex id: vertex id of the atom in the boost graph used to represent the molecular topology. This type is provided by the graph library used, boost graph in our case.
The aforementioned indices are used in the following data structures:
the order in which particles are created while building the graphs does not in general correspond to the ordering of atoms in the file. Whence:
The molecular covalent structure (MCS) of a biomolecule is a graph, possibly with several connected components. The MCS is created and then edited. Ultimately, a mapping between the topology and geometry (embedding) is realized.
Creating the MCS.
The CS data structure is represented as a boost subgraph. Such a graph contains vertices and edges. Each vertex (containing the information about a particle) is accessed via a so-called particle representation (particle rep), and an edge (modeling a covalent bond) via a bond representation or a pair of particle reps.
The MCS of a biomolecule is built upon the sequence of units (amino acids, bases) found in the input file.
In both cases, a post-processing step is performed to check that consecutive a.a. in the sequence specification are consecutive by ensuring that the resids are so.
Atoms and embedded atoms.
The covalent structure represents the graph of a molecular system, linking residues for proteins or nucleic acids. However, in a classical structure obtained by say X ray crystallography, selected atoms may be missing – e.g. hydrogen atoms or atoms located in flexible regions. Such are atoms are found in the covalent structure, but their geometry is not accessible. This motivates the following
Editing the MCS.
Once created the MCS undergoes the following editing steps:
Creating maps.
The following maps are then set to make it possible to connect the molecular graph with coordinates. Atomids may not be consecutive. To ease the processing, we define a consecutive version of atomids as follows:
The maps of interest are created in two steps:

Accessors for graphs and subgraphs.
As a graph, the MCS can be accessed and visited using iterators. Broadly, there are two main settings:
Other accessors.
The MCS data structure offers a panel of iterators, including:
To understand the various instantiations provided, let us start with the class used inside nodes of the graph data structure. This class is itself parameterized by the so-called particle traits:
Various particle traits are provided to accommodate annotation rich instantiations – file Molecular_covalent_structure_builder_instantiations.hpp :
Using the previous yields four lineages of instantiations, referred to as FIT, FIAT, HIT, and HIAT. Here is the lineage of instantiations using the FIT:
We have seen in the previous section that different applications use different types of atoms. This mechanism actually uses two levels:
We analyse these building blocks in the sequel.
The class SBL::CSB::T_Particle_info_traits< ParticleTraits > provides all the necessary types and operations depending on the model of ParticleTraits . Such a mechanism uses the C++ Partial Template Specialization : any structure can be used as a model of the concept ParticleTraits provided that the class SBL::CSB::T_Particle_info_traits is redefined for this particular model. The required operations depend on the context where it is used :
For any covalent structure, the requirements are given by the class SBL::CSB::T_Molecular_covalent_structure : SBL::CSB::T_Particle_info_traits , which must define two operators:
Less : comparator used to retrieve a vertex in the covalent structure from its info.
Printer : prints the information of the particle into a stream passed as argument.
For proteins and nucleic acids, the structure SBL::CSB::T_Particle_info_biomolecules wraps a Particle_type defined from the concept ParticleTraits. Usually, one can use the model SBL::Models::T_Atom_with_hierarchical_info_traits, that uses the Molecular_system atom type as Particle_type. This can be done by including the file "SBL/CSB/Particle_info_proteins.hpp".
SBL::CSB::T_Particle_info_traits must define three operators:
Finder : uses the covalent structure and biochemical data identifying a particle (chain identifier, residue sequence number, insertion code, atom name) to retrieve the corresponding vertex in the covalent structure;
Builder : builds an info from the relevant biochemical data (chain identifier, residue sequence number, insertion code, atom name, or a Molecular_system atom having all this data) and a terminal tag (positive if N-terminal, negative if C-terminal, null otherwise);
Aliases implements a dictionnary of aliases for the atom names in a protein, in order to have a unique atomic naming convention – see atom-naming-convention;
For other molecules, the requirements are given by the class SBL::IO::T_Molecular_covalent_structure_loader_from_MOL : SBL::CSB::T_Particle_info_traits must define the operator Builder ;
The default implementation provided uses internally a graph data structure to code particles and bonds, which comes with the following pros and cons:
Basically, the class SBL::CSB::T_Molecular_covalent_structure is a wrapper for a boost graph with accessors, modifiers and iterators over the graph. More precisely, it uses the boost subgraph data structure to offer the possibility of clustering the covalent structure, e.g by polypeptidic chains or by residues. There is a unique template parameter ParticleInfo representing the information attached to a particle, that is a simple string by default. An object of type ParticleInfo should be ordered, streamable and default constructible. Four main functionnality are available.
Building the covalent structure.
Adding a particle to the covalent structure is done using the method SBL::CSB::T_Molecular_covalent_structure::add_particle : this method takes a particle info representing that particle.
Adding a bond is done using the method SBL::CSB::T_Molecular_covalent_structure::add_bond : this method takes two vertices of the graph, and adds, if possible, an edge between the two vertices. It also takes optionnaly a bond type (simple by default, or double) to represent the covalence of the bond.
Mapping particleInfo to graph particleRep.
To retrieve particle reps from particleInfos, we define the map
Mapping particleReps to atomic coordinates. In the SBL, a geometric model is represented by a conformation that is a D-dimensional point consisting of the cartesian coordinates of the particles of a molecule. By default, the ith inserted vertex is mapped to the ith particle in the conformation.
Given an embedded vertex, it is then possible to access x, y and z coordinates from the covalent structure from the methods SBL::CSB::T_Molecular_covalent_structure::get_x, SBL::CSB::T_Molecular_covalent_structure::get_y and SBL::CSB::T_Molecular_covalent_structure::get_z .
Since a particle reps are in 0..n-1, with position the index in the molecular system as defined by the loader in the function
Mapping atom serial number to particle rep.
To locate a corresponding covalent structure node from a Molecular_system::Atom:
Iterating over the covalent structure.
It is possible to iterate over the particles, bonds, bond angles and torsion angles of the covalent structure. There are two modes of iteration : over all the entities, or over all the entities that are mapped with a geometric model. For example, if ones want to compute internal coordinates, it is only possible to do so from particles that are mapped. To switch between the modes, each iteration method has as argument an optional boolean tag (by default, it iterates over mapped particles).
Consider a set of particles associated with one or several connected components of the covalent structure. The subgraph induced by these particles is returned by SBL::CSB::T_Molecular_covalent_structure::get_sub_structure . The type of this graph is SBL::CSB::T_Molecular_covalent_structure . Note that stored information are not duplicated : modifying the sub-structure modifies also the parent covalent structure, and modifying the parent covalent structure modifies the sub-structure.
As an application, one can iterate on the individual molecules of a molecular system, as these correspond to the connected components of the covalent structure. To do so, one proceeds as follows :
(i) find the molecules using the method SBL::CSB::T_Molecular_covalent_structure::get_molecules which fills a container of particles, with one particle for each molecule;
(ii) for each found particle, create the subgraph containing that particle using the method SBL::CSB::T_Molecular_covalent_structure::get_molecule;
Displaying the covalent structure.
The covalent structure can be dumped in a file using Graphviz through the method SBL::CSB::T_Molecular_covalent_structure::print . For large systems (e.g. large proteins), the high number of particles, the Graphviz software may take a time to create the output. Also note that the neato command from Graphviz has a better rendering than the more classical dot command in this case.
 |
| Builders and the builder for biomolecules: the diamond pattern. The builder uses individual builders for proteins and nucleic acids, which themselves inherit common features from a generic builder for linear polymers. |
Building proteins and nucleic acids. To build the MCS of a biomolecule, one needs to know the sequences of residues: sequence of a.a. for a protein, and sequence of bases for a nucleic acid.
As noticed earlier, a given input file may contain a complex with both types of biomolecules. For this reason, the class SBL::CSB::T_Molecular_covalent_structure_builder_for_biomolecules is parameterized by two templates, namely one builder for proteins and one builder for nucleic acids.
The builder for biomolecules also contains the following enum to distinguish between both cases:
Upon processing a sequence (vector) of residue, the flag is properly positioned, so that the function make_residue() call the appropriate builder (for proteins or nucleic acids). As indicated below, the flag is set using the number of chars of the residue name.
The following is an excerpt from the file Core/Molecular_covalent_structure/include/SBL/CSB/Molecular_covalent_structure_builder_biomolecules.hpp
Required pieces of information. Independently from the biomolecular type, building a covalent structure requires three pieces of information:
File SBL/CSB/Molecular_covalent_structure_builder_generic.hpp.
SBL::CSB::T_Molecular_covalent_structure_builder_genericMolecular_covalent_structure_, SpecializedCSBuilder> : a generic addition of nodes / edges to a covalent structure assuming a polymer of units also called residues.
The SpecializedCSBuilder must provide 3 operations: make_first_residue(), make_residue(), make_last_residue().
In doing so, nodes and edges are added to a covalent structure passed as argument.
File SBL/CSB/Molecular_covalent_structure_builder_proteins.hpp. Specialized builder for proteins.
the class T_Molecular_covalent_structure_builder_for_proteins<Molecular_covalent_structure_> implements make_backbone_unit() (proteins: N-CA-C), make_first_residue(), make_residue(), make_last_residue() to build a polymer of amino acids.
The class SBL::CSB::T_Molecular_covalent_structure_builder_for_proteins is a functor building the covalent structure from an arbitrary data structure SBL::CSB::T_Molecular_covalent_structure_builder_for_proteins::Structure defining the polypeptidic chains and their residues. More precisely, a protein is represented by a mapping from a chain identifier to the chain itself, each chain being represented by a list of pairs (residue name, residue id). The graph of the covalent structure can be constructed at different scales : (0) with all atoms from all residues, (1) with only the heavy atoms, (2) with only the carbon alpha of each residue, or (3) with pseudo-atoms from the Martini coarse grain model. The scale factor is determined at the construction of the builder and is 0 by default. The only template parameter Covalentstructure is the covalent structure type.

File SBL/CSB/Molecular_covalent_structure_builder_nucleic_acids.hpp . Specialized builder for nucleic acids
the class T_Molecular_covalent_structure_builder_for_nucleic_acids<Molecular_covalent_structure_> implements make_backbone_unit(), make_first_residue(), make_residue(), make_last_residue() to build a polymer of bases.
File SBL/CSB/Molecular_covalent_structure_builder_biomolecules.hpp. The class T_Molecular_covalent_structure_builder_for_biomolecules<CovStructBuilderForProteins,CovStructBuilderForNucleicAcids> is parameterized by two specialized builders for proteins and nucleic acids respectively.
It contains two data members corresponding to these builders.
It main operator
processes a map, whose (key,value) pairs give access to a vector of residues for each chain. Upon processing a vector, the length of the name of the first residue is inspected, and used to decide which specialized builder must be called.
In practice, this class is instantiated with the two specialized builders, which are themselves instantiated with the same covalent structure:
This design makes it possible to create a graph containing different connected components for proteins and nucleic acids. It is also possible to edit this graph and create a covalent bond between both, if such a bond exists.
NB: Default_molecular_covalent_structure_builder is the builder to use in practice.
Since there is no particular difficulty nor operation for loading molecules from the MOL format, building the covalent structure from a MOL file is directly done in the loader SBL::CSB::T_Molecular_covalent_structure_loader_from_MOL .
The class SBL::CSB::T_Molecular_covalent_structure_loader is the orchestra conductor.
template <class Molecular_covalent_structure_builder_>
class T_Molecular_covalent_structure_loader : public T_Molecular_system_loader<>{
};
Step 1. creation of the covalent structure.
Step 1: covalent graph creation. The constructor
uses
The add_particle comes from T_Molecular_covalent_structure<ParticleInfo>::add_particle(const Particle_info& info), which does 2 critical things:
creation of an entry <particle_info, Particle_rep> in the map m_particleInfo_to_particleRep NB: this is critical since when setting coordinates with the function include/SBL/IO/Molecular_covalent_structure_loader.hpp make_covalent_structure_conformation_mapping(model, mcs);
NB2: make_covalent_structure_conformation_mapping() now adds entries to the map atomSerialNumber_to_particleRep for each atom in the molecular system
Step 2: setting coordinates.
The critical function is
since it returns the graph node to which the coords will be associated. this graph node must have been set by main step 1.
File SBL/IO/Molecular_covalent_structure_loader.hpp.
the main class, which creates the covalent structures, and associates coordinates.
The loading of PDB and mmCIF files is described in the packages ESBTL and Molecular_system.
Practically, the following file formats are used:
The (legacy) PDB format: the reference format for biomolecules whose structure has been resolved and are deposited in the Protein Data Bank ( PDB website).
The mmCIF file format.
As noted above, loaders for MCS depend on loaders for molecular systems from the package Molecular_system. Such loaders use the libcifpp library.
Loader from PDB/mmCIF files.
The class SBL::IO::T_Molecular_covalent_structure_loader is a loader as defined in the Module_base package : it loads an input PDB/mmCIF, uses a builder to build a covalent structure, and maps the vertices of the covalent structure to the loaded atoms by order of their atomic sequence identifier.
The following comments are in order:
Water molecules are not loaded by default. To change this behaviour, one can use the methods SBL::IO::T_Molecular_covalent_structure_loader::set_loaded_water. Then, water molecules are added as independent connected components in the covalent structure graph.
Hetero atoms are not loaded by default. In the future, the flag SBL::IO::T_Molecular_covalent_structure_loader::set_loaded_hetatoms will be used to load them. Note that for hetero atoms which are not monoatomic ions, the connectivity is unknown.
As note above, disulfide bonds are added to the built covalent structure, see the package Pointwise_interactions. This feature is available by default but can be turned off using the method SBL::IO::T_Molecular_covalent_structure_loader::set_without_disulfide_bonds .
It is possible to change the maximal authorized length of the bond between the C of a residue and the N of the next residue using the method SBL::IO::T_Molecular_covalent_structure_loader::set_max_bond_distance.
Specific atomic naming conventions.
Loader from MOL files.
The class SBL::IO::T_Molecular_covalent_structure_loader_from_MOL is a loader as defined in the Module_base package : it simply loads an input MOL file, and directly builds the covalent structure from the loaded data and maps the vertices to the laoded particles. It has a unique template parameter MolecularCovalentStructure , that is the representation of the covalent structure, by default SBL::CSB::T_Molecular_covalent_structure <> (see package Molecular_covalent_structure)
It is possible to set the coarse level used by the builder to create the covalent structure. This class has four template parameters, all having a default type :
MolecularCovalentStructure : representation of the covalent structure, by default SBL::CSB::T_Molecular_covalent_structure <> (the parameter ParticleInfo is by default a simple string, see package Molecular_covalent_structure)
MolecularCovalentStructureBuilder : builder of the covalent structure, by default SBL::CSB::T_Molecular_covalent_structure_builder_for_proteins < MolecularCovalentStructure >,
Special residues can be present in biomolecules. In general, these are incorporated artificially for labeling purposes in crystallographic studies – molecular replacement. For some of these special residues, we provide the possibility of either loading them directly or replacing them with their natural counterparts.
Currently the list of supported residues is the following:
For proteins only.. We provide the possibility of handling unsupported special residues by loading them into the covalent structure as unknown residues (name: UNK) with a placeholder covalent structure matching that of alanine. Indeed, since the topology of UNK is unknown, one cannot build the corresponding covalent subgraph. The benefit of doing so is to enable the creation of the MCS, e.g. to enable statistical analysis of internal coordinates.
This design choice has two consequences:
These options are passed on the command line as –residue-natural and –residue-unknown – See Figure below.
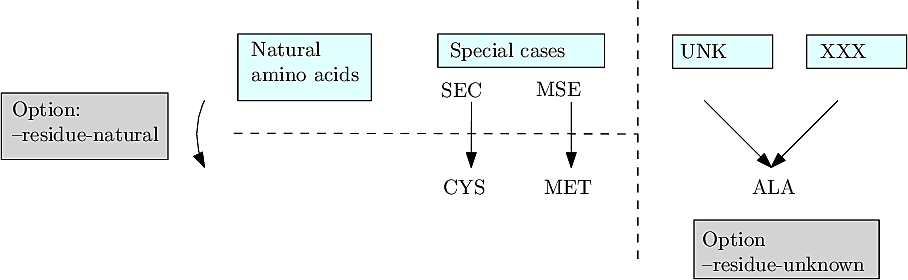 |
| Options to handle non standard residues when building a covalent structure. |
Manipulating molecular coordinates (package Molecular_coordinates) or computing potential energies (package Molecular_potential_energy) benefits from a canonical representation of internal coordinates.
The method SBL::CSB::T_Molecular_covalent_structure::get_canonical_rep returns the cannonized representation of a bond, a bond angle or a dihedral angle.
Canonical representation of a bond. Such a representation is obtained by ordering its two vertices – that is the particle_rep used to represent the vertices.
Canonical representation of a valence angle. Such a representation is obtained by ordering the first and third vertices – since the middle one is imposed.
Canonical representation of a dihedral angle. This case covers two sub-cases (see also the package Molecular_coordinates):
So far, we have described a covalent structure based on a graph data structure.
When the MCS is used to compute the potential energy and the gradient of the energy of a large number of conformations, it becomes necessary to optimize its traversal – and the calculation of internal coordinates. The base data structure uses a graph for representing it, so that it is easy to visit particles and bonds. However, the following comments are in order:
For these reasons, two covalent data structures are provided:
The class SBL::CSB::T_Molecular_covalent_structure_optimal< ParticleInfo > is an optimized version of the covalent structure data structure. In particular, all entities (particles, bonds and angles) that are used in the computation of the energy are stored in a vector, ensuring a constant time access each time.
In order to use this class, once has to first build a covalent structure with the class SBL::CSB::T_Molecular_covalent_structure, then use the class SBL::CSB::T_Molecular_potential_energy_structure_parameters_traits_optimal to build the optimized version of the covalent structure data structure. See the package Molecular_potential_energy for examples and more details.
The following example shows how to load a PDB file and build the corresponding covalent structure at a given scale. It then output a dot file in the Graphviz format to check the covalent structure.