|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
![]()
Authors: F. Cazals and T.O'Donnell
This package provides tools to generate or modify tripeptide segments in protein structure, using the so-called Tripeptide Loop Closure (TLC). These tools encompass low level C++ code, as well as the application 
Classical TLC.. Mathematically, consider a tripeptide whose internal coordinates (bond lengths 



Solving the problem requires finding the real roots of a degree 16 polynomial, which also means that up to 16 solutions may be found [163], [67].
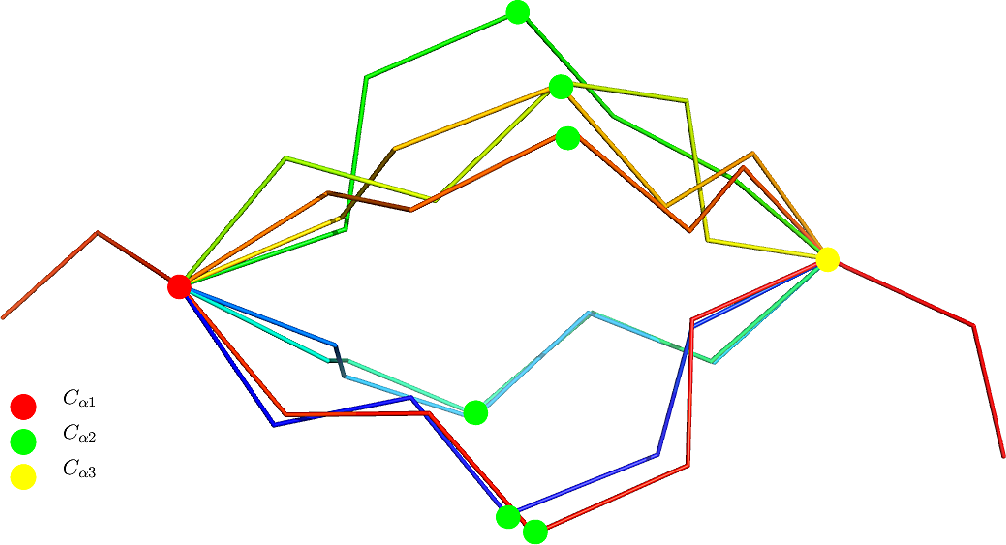 |
| TLC: example reconstructions. |
TLC with gaps. A generalization of the classical TLC consists of considering three amino acid which are not not contiguous along the backbone (Fig. fig-TLCG-example).
This case is of interest in the presence of three linkers enclosing two rigid SSE. Mathematically, this is akin to the original problem, with the rigid blocks modeled as fictitious bonds separating the amino acid (Fig. fig-TLCG-example for one example).
TLCG: example reconstructions sandwiching a beta sheet. PDBID 1vfb, chain C. The three amino acid defining the tripeptide are:    |
Our implementation follows [67], except for numerics and the handling on internal coordinates other than the dihedral angles.
TLC and internal coordinates. Solving a particular TLC problem puts the focus on dihedral angles, so that that there are three options to handle the other internal coordinates (bond length and valence angles):
#6 bond lengths from CA1C1 to N3CA3 1.33 1.52 1.45 1.33 1.52 1.45 #7 bond angles from N1CA1C1 to N3CA3C3 1.95 2.05 2.01 1.95 2.05 2.01 1.95 #Omega torsion angles 3.14 3.14
Algorithm. First, the constraints and position of anchors around the loop closure are extracted. These atoms are 
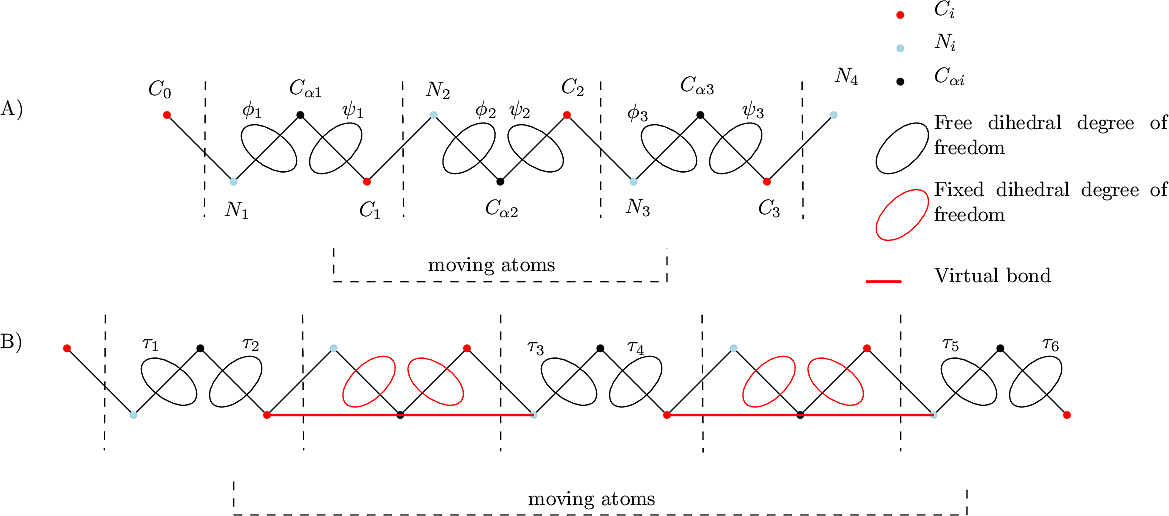 |
Tripeptide: atoms and degrees of freedom used for loop closure. (A) Classical tripeptide loop closure(TLC), the six dihedral angles represented correspond to the degrees of freedom used to solve the problem. (B) In tripeptide loop closure with gaps(TLCG), the dihedral degrees of freedom  |
TLC constraints. Consist of the positions of anchors (2 legs i.e. four points), and the remaining internal coordinates (6+7+2).
TLC constraints and associated loader.
The constraints used to solve TLC are (i) the positions of the anchors (left and right legs), and (ii) the remaining internal coordinates, namely bond lengths (6 values), valence angles (7 values), and torsion angles (2 values, omega angles).
The main classes defining and loading these constraints are
TLC options. The options used by TLC encompass
These options are managed in the following class, which also provides the associated command line parsing:
Main algorithm. The main algorithm takes as input a protein representation and the TLC options. It consists of the following steps:
The corresponding class, which is parameterized by a Traits class defined the required types, is:
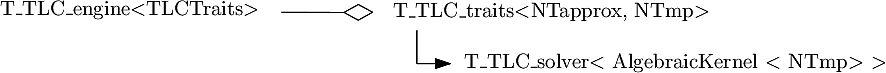 |
| Main classes. NTmp and NTapprox respectively stand for NumberTypeMultiPrecision (typically CGAL::Gmpfr) and NumberTypeApprox (typically double). The former is used to solve the TLC degree 16 algebraic system, and the latter is used to return solutions. |
Application. The final application undertakes the following steps
Algebra.
Algebra. Four classes following the steps of the algorithm are provided:
These three classes have an algebraic kernel as single template argument(default CGAL::Algebraic_kernel _d_1<CGAL::Gmpq>>). This kernel defines the polynomial type in step 1, the solver in step 2 and the Roots Number type in step 3. These three classes are implemented in a fourth interface class SBL::CSB::T_Tripeptide_loop_closure were all the steps are performed using the same kernel.
Robustness. The numerical stability of an algorithm is key to its robustness [34]. The aforementioned three steps use two number types:


Using these, the three steps go as follows:
Utilities. A separate class (SBL::CSB::Tripeptide_loop_closure_utilities) with a number of useful low level static methods, in particular matrix operation functions using arrays.
A number of operations on internal coordinates from the package Molecular_coordinates are used:
This package provides the executable
More specifically, the main options of


The following comments are in order:



The format for user provided internal coordinates is as follows:

#6 bond lengths from CA1C1 to N3CA3 1.33 1.52 1.45 1.33 1.52 1.45 #7 bond angles from N1CA1C1 to N3CA3C3 1.95 2.05 2.01 1.95 2.05 2.01 1.95 #Omega torsion angles 3.14 3.14
We note in passing that the following default values are used:
#Bond lengths ac cn na ac cn na 1.52 1.33 1.45 1.52 1.33 1.45 #Bond angles nac acn cna nac acn cna nac 1.947 2.050 2.093 1.947 2.050 2.093 1.947 #Omega torsion angles 3.14159265359 3.14159265359
Assume that a number of solutions have been generated. For example, consider the 8 solutions of the following call, corresponding to changes in a loop of an antibody:
(old) sbl-tripeptide-loop-closure.exe --filename 1vfb.pdb --chainid A --resids 11-13-15 -o solutions (new) sbl-tripeptide-loop-closure.exe -f 1vfb.pdb --load-chains A --resids 11-13-15 --odir solutions
We provide the script $SBL_DIR/scripts/vmd/load-pdbs.vmd, which can be used as follows
vmd -e /path/to/vmd/script/load-pdbs.vmd -args original/pdb/file.pdb solutions*.pdb
In the context of the previous section, all input PDB files can be passed at once to pymol:
pymol input-file.pdb solutions*.pdb
The SBL provides VMD, PyMOL, and Web plugins for sbl-tripeptide-loop-closure.exe. Launch the VMD plugin from the SBL catalog under the Extensions menu, start the PyMOL plugin by running sbl_tripeptide_loop_closure in the PyMOL command line, and open the Web plugin from the SBL web plugins launcher page (served locally via sbl-web-plugins-launcher). All three plugins expose the same GUI and capabilities.
 |
| sbl-tripeptide-loop-closure plugin user interface. The user interface for the sbl-tripeptide-loop-closure plugin. The typical workflow is: Step 1: Provide input file. Step 2: Specify the chain id and the three residue ids. Step 3: Choose an output directory (optional, a default is provided). Step 4: Launch the computation. Step 5: Inspect the conformation solutions in the viewer. Step 6: (Optional) Use the update panel to select separate solution to view. |
See the following jupyter notebook:
import os
import nglview as nv
The options of sbl-Tripeptide-loop-closure are:
--filename, Input pdb file(must have .pdb suffix}
--chainid, id of the chain in which the loop is found
--resids, resids(idn) concateneted with eventual insertion codes(cn) in the format id1c1-id2c2-id3c3
--precision-factor, Multiplicative factor (double \eql{>1}) for the number of bits in the mantissa
--data-internals, Use the internals found in data. (Nb: all backbone atoms in the loop must be present in the file)
--standard-internals, Use standard internal values
--user-defined-internals, specify internal value constraints from a file
--directory-output, Folder where output files will be stored
--output-prefix, prefix for output files
--help, print the above option details#create output folder
outputfolder="results"
if not os.path.exists(outputfolder):
os.mkdir(outputfolder)
#Generate Tripeptide loop closure solutions
os.system("sbl-tripeptide-loop-closure.exe --filename data/1vfb.pdb --chainid A --resids 11-13-15 --output-prefix data_ --directory-output results")
os.system("sbl-tripeptide-loop-closure.exe --filename data/1vfb.pdb --standard-internals --chainid A --resids 14-15-16 --output-prefix std_ --directory-output results")
os.system("sbl-tripeptide-loop-closure.exe --filename data/1vfb.pdb --user-defined-internals constraints.txt --chainid A --resids 14-15-16 --output-prefix specified_constraints_ --directory-output results")
Visualize original file:
view = nv.show_file("data/1vfb.pdb")
view
Visualize data extracted internals second solution:
view = nv.show_file("results/data_1vfb-solution-2.pdb")
view
Visualize standard internals second solution:
view = nv.show_file("results/std_1vfb-solution-2.pdb")
view
Visualize user defined internals second solution:
view = nv.show_file("results/specified_constraints_1vfb-solution-2.pdb")
view