|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
Authors: F. Cazals and T. Dreyfus and D. Mazauric and C. Roth
![]()
This package provides methods to compare, from a geometric/structural standpoint, two ensembles of conformations.
Consider two conformational ensembles, also called samplings, as defined in section Conformational ensembles – aka samplings . Let 
This package offers methods two compare these two ensembles in different ways:
computing the Hausdorff distance between ensembles
These functionalities are provided in the program 
A standard problem in analyzing ensembles of conformations consists of comparing two such ensembles. For example, one may wish to compare two sets of local minima, or two sets of structures selected for their relevance in a particular context.
To perform such an assessment, consider two sets of conformations, denoted for convenience 

We do so by computing a minimal spanning forest of a weighted complete bipartite graph induced by the two sets 


Note that the following pieces of information are returned:
High level statistics: the sum of weights of oriented arcs.
Low level statistics: the list of oriented edges and their weight that is a list of triples.
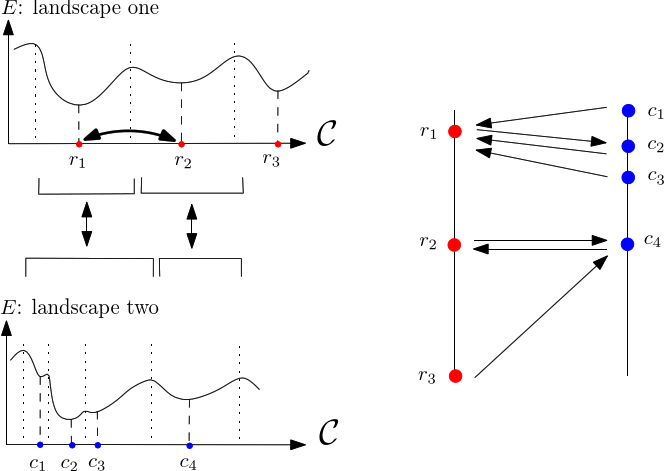 |
| Comparing sets of minima using a Minimum Oriented Spanning Forest (MSF) Each conformation from a sampling picks the nearest neighbor in the second set. Note that the process is not symmetric:    |
To get a global criterion, we introduce the so-called Hausdorff distance, a 
Consider the previous sets of conformations

We then report the following triple:

Note that the third entry in Eq. (eq-hausdorff-triple) is the so-called one-sided Hausdorff distance between
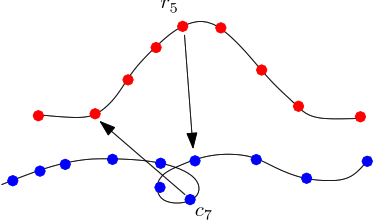 |
| One sided Hausdorff distance Given two sets of conformations, one seeks for each of them the sample located furthest in the second sampling. Maximizing this distance yields the one sided Hausdorff distance. |
Given two samplings

the symmetric difference 


Note that the previous set operations are performed up to a user-specified tolerance 
The input is two conformational ensembles of the same molecule, and can be provided with several format:
6 x11 y11 z11 x12 y12 z12 6 x21 y21 z21 x22 y22 z22 6 x31 y31 z31 x32 y32 z32 ...
All comparisons are optional, so that they have to be specified in the command-line. For example, running Hausdorff comparison is done using the option –Hausdorff. Thus, a calculation is launched as follows:
> sbl-conf-ensemble-comparison-lrmsd.exe --points-file data/bln69_sampling.txt --points-file data/bln69_10_lowest_minima.txt --Hausdorff --symmetric-difference --identity-threshold 0.01 --exact-search --directory results --verbose --output-prefix --log
| File Name | Description |
| BLN69 conformations file | Sampling done from sbl-landexp-hybrid-BH-TRRT-BLN.exe |
| BLN69 conformations file | Ten lowest local minima of BLN69 obtained by an external sampling method |
| Preview | File Name | Description |
| General: log file | ||
| Log file | Log file containing high level information on the run of | |
| Module MSF analysis: Minimum spanning forest | ||
| Conformation matches text file | List of edges of the MSF with the distance between the selected conformations | |
| Module symmetric difference analysis: intersection and symmetric difference | ||
| Conformation matches text file | List the pair of indices of conformations that match | |
It is easily shown that an optimal subset of edges induces a forest; i.e., a subgraph without cycles. A simple algorithm consists of choosing, for every node, the incident edge with the smallest weight. This strategy corresponds to the first step of 
Two strategies are offered to compute the nearest neighbor of each sample:
Brute force: a linear scan is used to find the exact nearest neighbor.
Approximate: data structures to perform approximate nearest neighbor searches are used, see package Spatial_search
The nearest neighbor of each sample is computed during the MSF calculations. This information is used to compute the Hausdorff triple of Eq. eq-hausdorff-triple.
The programs of Conformational_ensemble_comparison described above are based upon generic C++ classes, so that additional versions can easily be developed.
In order to derive other versions, there are two important ingredients, that are the workflow class, and its traits class.
T_Conformational_ensemble_comparison_traits:
T_Conformational_ensemble_comparison_interface_workflow: