|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
Authors: F. Cazals and T. Dreyfus and A. Lheritier and N. Malod-Dognin
![]()
A tenet of Science is the ability to reproduce the results, and a related issue is the possibility to archive and interpret the raw results of (computer) experiments. This package presents an elementary python framework focusing on the archival and the analysis of raw data, with three goals in mind:
To see how these goals are met, consider a computing pipeline consisting of raw data generation, raw data parsing, and data analysis i.e. graphical and statistical analysis. PALSE addresses these last two steps by leveraging the hierarchical structure of XML documents.
More precisely, assume that the raw results of a program are stored in XML format, possibly generated by the serialization mechanism of the boost C++ libraries.
For raw data parsing, PALSE imports the raw data as XML documents, and exploits the tree structure of the XML together with the XML Path Language to access and select specific values. For graphical and statistical analysis, PALSE gives direct access to ScientificPython, R, and gnuplot.
In a nutshell, PALSE combines standards languages (python, XML, XML Path Language) and tools (Boost serialization, ScientificPython, R, gnuplot) in such a way that once the raw data have been generated, graphical plots and statistical analysis just require a handful of lines of python code.
The framework applies to virtually any type of data, and may find a broad class of applications.
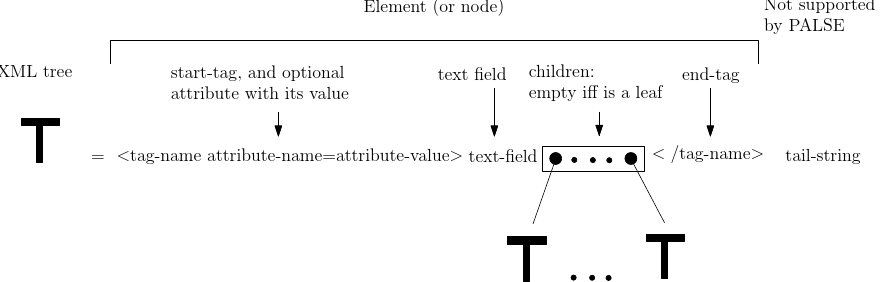
XML provides a hierarchical representation of a document, which may be seen as a rooted ordered tree of nodes called Elements – see Fig. fig-palse-xml-tree. Each Element is characterized by a tag-name, and possibly contains attributes, a text field and a number of child Elements — there is also an optional tail string which is not supported by PALSE):

In the sequel, by data value, we refer either to the value of an attribute or of a text field.
An Element may be also embedded in a namespace, that is represented by a pair (prefix, uri). The uri is a string of characters identifying a name or a resource, and the prefix is a simple name associated to the uri. Thus, if an Element
However, in the XML tree, all prefixes are replaced by their corresponding uri, so that the tag name of the Element
|
|---|
Given an XML document, the XPath language has been developed to address specific parts of this document – see the XPath documentation page. Recall that an XML document is represented as a rooted ordered tree: a XPath query always specifies a path to one or more of its Element(s).
An XPath is specified with sequences of tags such as 


To define relative searches, let the contextual element be the current element in a path:



The following are absolute searches i.e. start the search at the root:




![$tag[@att]$](form_1003_dark.png)
![$tag[@att='val']$](form_1004_dark.png)

![$.//tag[@att]$](form_1006_dark.png)
PALSE assumes that a given (computer) experiment yields one file storing the results. To make these data perennial and foster their availability, PALSE uses XML, since this language is a standard one, and most importantly, provides an abstract structure amenable to high-level querying and filtering operations.
In this section, we present a short example in which one computes various statistics from a Database storing surface areas and volumes of atoms in molecules, computed using tools from the package Space_filling_model_surface_volume.
In PALSE, each result file is loaded into an XML ETree, so that one gather data by traversing paths in this tree.
Consider for example a case where one has generated an XML file to describe the volume of the restrictions of the atoms to their Voronoi cells. First, to inspect the hierarchy embedded in an XML file, one can use the method SBL::PALSE::PALSE_xml_DB::get_common_hierarchy_as_xml for generating a string representation of this hierarchy:
It is also possible to access to the list of all paths from the root of XML files rather than the XML format, using the method SBL::PALSE::PALSE_xml_DB::get_common_hierarchy_as_list :
The previous also shows that values can be accessed in two ways from an XML file, that is:
This duality is especially relevant for serialized containers such as those from the STD library. On the previous example, one gets:
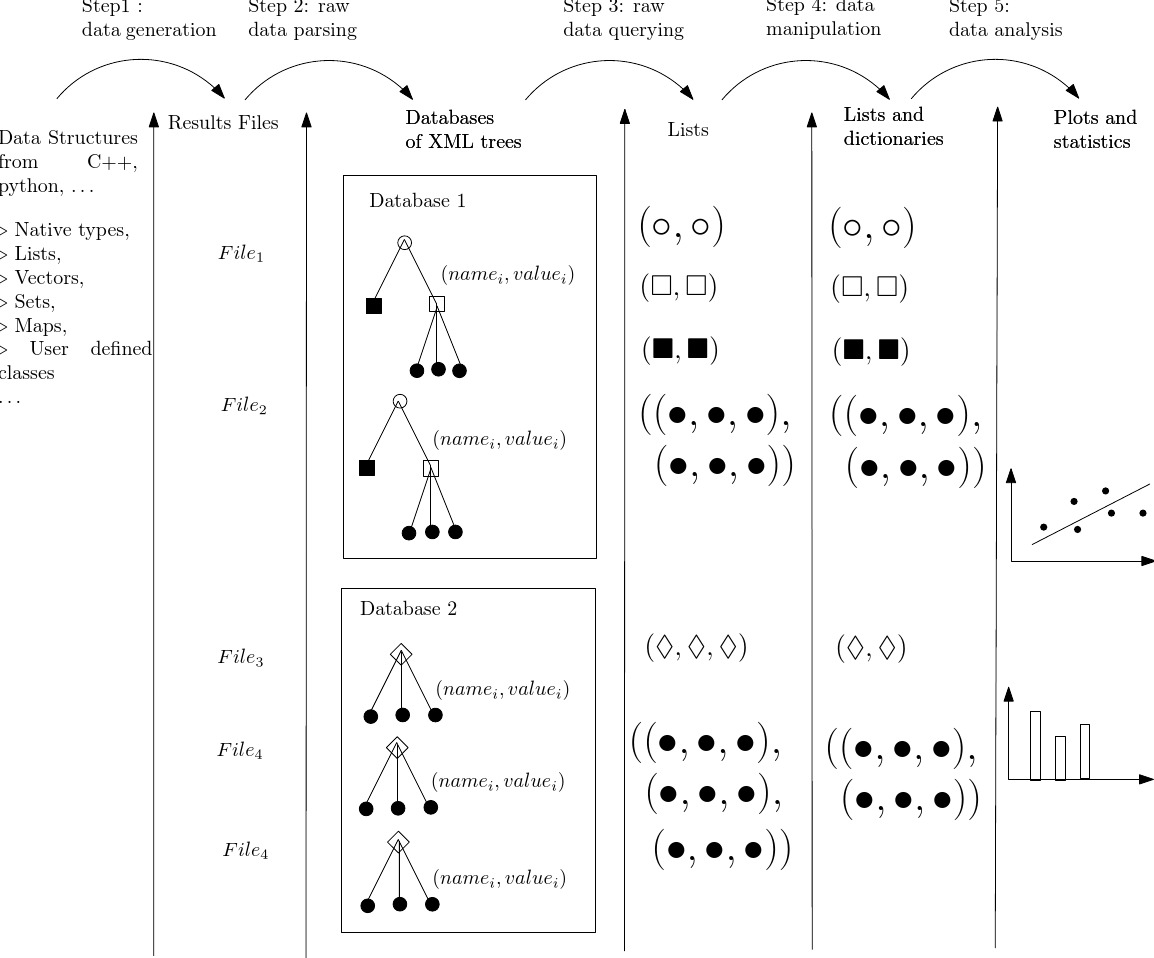
This section describes more precisely the five different steps used in the PALSE methodology. These five steps are summarized on Fig. fig-palse-workflow.
|
|---|
By raw data we refer to the results of some (computer) experiment—PALSE can be used to handle data generated by any device, or even manually archived. Archives and their (de-)construction is a tenet of PALSE following the Boost library serialization engines. As specified in the Boost documentation,
we use the term "serialization" to mean the reversible deconstruction of an arbitrary set of C++ data structures to a sequence of bytes. Such a system can be used to reconstitute an equivalent structure in another program context.
That is, an archive refers to a specific rendering of the aforementioned sequence of byte, and PALSE takes as input XML archives.
For computer experiments, the generation of XML data naturally depends on the programming language used, two of them being of particular interest: C++ and python.
In C++, the Boost Serialization tools accommodate the native C++ types and the data structures of the Standard Template Library.
In python, the recursive structure of dictionaries makes conversion of dictionary into an XML tree (and vice-versa) a trivial task.
Consider a set of XML archives, one per (computer) experiment. PALSE imports each such file as an XML tree. A database corresponds to a set of isomorphic trees corresponding to experiments generated with varying parameters. Therefore, several databases may be used to accommodate several sets of parameters, or to store results from different sources. The creation of the trees from the files is delegated to the lxml library (lxml is an easy-to-use library for handling XML and HTML in Python, see the documentation page.
Given the XML trees stored in database(s), the retrieval of the values of interest for graphical and statistical analysis combines features of the XML and XPath, as discussed in section The XPath Query Language .
Functions offered in PALSE . The three XPath modes discussed in section The XPath Query Language are used by PALSE functions to query the database, in order to retrieve selected Elements or their data values (either from text fields or attributes):
While queries on data values return directly a list that can be treated with simple statistical tools, queries on Elements return a list that can be used to filter even more the Elements. Given a list of Elements 




We also note that upon calling a function returning Elements, the selection can be converted into strings thanks to the function SBL::PALSE::PALSE_xml_DB::get_data_values_from_elements (
PALSE using BioPython : BioPALSE. PALSE offers also functionality handling PDB files treated by the BioPython module. There are three main functionality allowing to convert the PDB files into XML trees:
The data collected by the previous step typically need to undergo processing before being amenable to analysis. Since the previous step supplies python lists of strings, PALSE provides mechanisms to select, sort, and convert these lists into lists of elementary types. Furthermore, PALSE provides tools for combining lists into dictionaries, for further filtering using regular expressions (for strings) or lists of allowed keys or values.
The lists just produced are ready for graphical and statistical analysis. PALSE encapsulates functionalities from SciPython and R for computing Pearson, Spearman or Kendall's tau correlations between the collected values, as well as Mann-Whitney rank-sum test. More generally, any package interface with python can be used, e.g. gmpy if unlimited-precision integers / rationals / floats must be used to ensure numerical correctness, or gnuplot to generate eye-candy plots and charts, etc.
//nvp for name-value-pair: allows to attribute a name to a
//serializable value. In this context, it is used for decomposing the
//serialization of a data structure
#include <boost/serialization/nvp.hpp>
//type of the archive file of the serialization: here, it is an output
//xml file (we will save the serialization in this file)
#include <boost/archive/xml_oarchive.hpp>
#include <fstream>
#include <iostream>
//Small data structure that we want to serialize
struct Data_type
{
int index;
};
//Definition of the serialize method for our small data structure. It
//takes an archive and load or save the serializable fields of the data
//structure, as described in the method. The symbol "&" means that the
//operation is the same for loading or saving the data structure.
namespace boost
{
namespace serialization
{
template <class Archive>
void serialize(Archive& ar, Data_type& data, const unsigned int version)
{
ar & boost::serialization::make_nvp("index", data.index);
}
}
}
//The main method creates an object of type Data_type, and then save
//it in a serialized xml archive.
int main()
{
Data_type data; data.index = 1;
//first, open the file, then open the file as an output xml archive.
std::ofstream out("test.xml");
boost::archive::xml_oarchive* ar = new boost::archive::xml_oarchive(out);
//save the data in the archive
*ar << boost::serialization::make_nvp("data", data);
//first close the archive, then close the file (the order is
//important, since some additional information are dumped in the file
//when closing the archive)
delete ar;
out.close();
return 0;
}
For our application, each XML file generated contains, amongst other things, the surface area ( <total_area></total_area>) and the volume ( <total_volume></total_volume>) associated with each atomic restriction.
The attached jupyter notebooks illustrates various features of PALSE to extract the following statistics:
For the details, check out:
We use the application Space_filling_model_surface_volume, see https://sbl.inria.fr/doc/Space_filling_model_surface_volume-user-manual.html, to compute surface areas and volumes of molecules.
Note that for each input PDB file, we define one space filling model for two different expansion radii:
The output files for r=0 and r=1.4 a dumped into two distinct directories. While this is done manually here, the process can be automated using the Batch_manager, see https://sbl.inria.fr/doc/Batch_manager-user-manual.html
import re #regular expressions
import sys #misc system
import os
import pdb
import shutil # python 3 only
exe = "sbl-vorlume-pdb.exe"
odir = "results"
idir = "data"
files = ["1avw.pdb", "1brs.pdb"] #, "1dfj.pdb"]
radii = [0, 1.4]
def compute_surfaces_volumes_of_pdb_files():
for radius in radii:
odir = "sbl-vorlume-pdb-data-radius%s" % radius
os.system( ("mkdir %s" % odir))
for ifile in files:
cmd = "%s -f %s/%s --directory %s --verbose --output-prefix --log --radius %s --report-options" % (exe, idir, ifile, odir,radius)
print
os.system(cmd)
compute_surfaces_volumes_of_pdb_files()
As a first application of PALSE, we compute the average volume of atomic restrictions for the SAS model. This involves the following steps:
from SBL import PALSE
from PALSE import *
def ave_volume_atoms():
database = PALSE_xml_DB()
database.load_from_directory("sbl-vorlume-pdb-data-radius1.4",".*volumes.xml")
volumes = database.get_all_data_values_from_database("restrictions/item/total_volume", float)
volumes = PALSE_DS_manipulator.convert_listoflists_to_list(volumes)
return sum(volumes)/float(len(volumes))
print("Average volume of atomic restrictions:", ave_volume_atoms())
The following comments are in order:
the search path does not contain the global tag
If the database is partitioned over several subdirectories, it is possible to recursively load all the XML files using the method load_from_directory(input_dir, regex, True)
One XML file listing several volumes, and a database possibly containing several XML files, the method get_all_data_values_from_database always returns a list of lists. If one does not want to discriminate in between the different XML files, the method convert_listoflists_to_list returns a unique concatenated list from an input list of lists.
In the example above, the files involved in the database do not obey any specific ordering. As explained below, the function database.sort_databases can be used to sort these files based on a regular expression acting on their names.
As a second example, we replicate the previous analysis, distinguishing the two radii used.
This requires two features:
adding the option --report-options to the run of the SBL executable, allowing to report an XML file listing all the options used for the calculations;
sorting the database of loaded XML files w.r.t a given option using the method PALSE_xml_DB::sort_databases.
def exple_average_volume_by_increasing_radius():
database = PALSE_xml_DB()
database.load_from_directory("sbl-vorlume-pdb-data-radius0",".*volumes.xml")
volumes = database.get_all_data_values_from_database("restrictions/item/total_volume", float)
volumes = PALSE_DS_manipulator.convert_listoflists_to_list(volumes)
print("Average restriction volume without added radius: ", sum(volumes)/float(len(volumes)))
database = PALSE_xml_DB()
database.load_from_directory("sbl-vorlume-pdb-data-radius1.4",".*volumes.xml")
volumes = database.get_all_data_values_from_database("restrictions/item/total_volume", float)
volumes = PALSE_DS_manipulator.convert_listoflists_to_list(volumes)
print("Average restriction volume with added radius 1.4 A: ", sum(volumes)/float(len(volumes)))
exple_average_volume_by_increasing_radius()
@Tom: we do not sort anymore !???
The following comments are in order:
The sort is done over strings by default. To change this behaviour, the second positional argument (or the type argument) can be set to any comparable type.
The XML archives containing all the options of a run of a SBL executable always terminate by "__options.xml". However, if for any reason the files listing the options are terminated differently, the third positional argument (or the options_suffix argument) can be set to any file suffix.
It is also possible to analyze specific elements amongst the data, such as the volume of a specific residue type, or the volume of an residue identified by its sequence number. To do so, three steps are required :
gathering all elements in the database at the desired level; for example, if the analysis undertaken focuses on residues, one has to gather all elements corresponding to residues; this is done with the method PALSE_xml_DB::get_all_elements_from_database that returns one list of elements for each dataset in the database;
filtering elements matching a given requirement; for example, assume that one wishes to select elements corresponding to residues of type "GLY", or only in the chain "A"; this is done through the method PALSE_xml_DB::filter_elements_by_data_values_equal_to that returns a filtered sublist of elements from the input list of elements;
accessing desired values from the filtered elements; for example, for each filtered residue, one may want to access its identifiers, volume and area; this is done with the method PALSE_xml_DB::get_all_data_values_from_elements that returns a list of values matching the input XPath query for each element in the input list of elements;
The following script shows how to print the average area for all non-buried glycines using the three methods:
def exple_average_area_specific_residue_type():
database = PALSE_xml_DB()
database.load_from_directory("sbl-vorlume-pdb-data-radius1.4",".*volume.xml")
residues = database.get_all_elements_from_database("residues/item")[0]
gly_residues = database.filter_elements_by_data_values_equal_to(residues, "residue_name", "GLY")
areas = database.get_all_data_values_from_elements(gly_residues, "area", float)
areas = [x for x in PALSE_DS_manipulator.convert_listoflists_to_list(areas) if x > 0]
print("Average residue free surface area of GLY with added radius 1.4 A: ", sum(areas)/float(len(areas)))
exple_average_area_specific_residue_type()
It is also possible to compute the volume statistics of atoms sharing a common property. For example, one wants to know the average volume of carbons depending on the residue containing it. The python script is similar to the one presented in section Example Python Script : Average Volume of Atoms, but one has to collect separately all the carbons of each residue type instead of just collecting all the atoms together. This is done in three steps:
This is done with the following python script:
def exple_average_volume_atom_property():
database = PALSE_xml_DB()
database.load_from_directory("sbl-vorlume-pdb-data-radius1.4",".*volumes.xml")
particles = database.get_all_elements_from_database("restrictions/item")
particles = PALSE_DS_manipulator.convert_listoflists_to_list(particles)
residue_names = database.get_all_data_values_from_elements(particles, "particle/residue_name")
residue_names = sorted(set(PALSE_DS_manipulator.convert_listoflists_to_list(residue_names)))
for resname in residue_names:
particles_of_residue = database.filter_elements_by_data_values_equal_to(particles, "particle/residue_name", resname)
carbons_of_residue = database.filter_elements_by_data_values_equal_to(particles_of_residue, "particle/atom_name", "C")
volumes = database.get_all_data_values_from_elements(carbons_of_residue, "total_volume", float)
volumes = PALSE_DS_manipulator.convert_listoflists_to_list(volumes)
if len(volumes) > 0:
print("Average volume of carbons in %s" % resname, sum(volumes)/float(len(volumes)))
else:
print("Average volume of carbons in %s" % resname, 0)
exple_average_volume_atom_property()
PALSE offers also the possibility to plot simple diagrams as 2D plots or histograms. For example, it is possible to plot the distribution of the volumes of the residues. To do so, the following steps are required:
collecting all the particles in the database per input XML file,
for each XML file, (i) collecting all the existing residues ids from the particles, (ii) classifying the particles per residue id, and (iii) summing the volumes of the particles of each residue,
plotting the histogram of the volumes of the residues.
This is done with the following python script:
def distribution_residue_volumes():
database = PALSE_xml_DB()
database.load_from_directory("sbl-vorlume-pdb-data-radius1.4",".*volumes.xml")
residue_volumes = []
particles = database.get_all_elements_from_database("restrictions/item")
for particles_per_file in particles:
chain_identifiers = database.get_all_data_values_from_elements(particles_per_file, "particle/chain_identifier")
chain_identifiers = sorted(set(PALSE_DS_manipulator.convert_listoflists_to_list(chain_identifiers)))
for chain in chain_identifiers:
particles_per_chain = database.filter_elements_by_data_values_equal_to(particles_per_file, "particle/chain_identifier", chain)
residue_sequence_numbers = database.get_all_data_values_from_elements(particles_per_chain, "particle/residue_sequence_number", int)
residue_sequence_numbers = sorted(set(PALSE_DS_manipulator.convert_listoflists_to_list(residue_sequence_numbers)))
for i in residue_sequence_numbers:
particles_of_residue = database.filter_elements_by_data_values_equal_to(particles_per_chain, "particle/residue_sequence_number", str(i))
volumes_of_particles = database.get_all_data_values_from_elements(particles_of_residue, "total_volume", float)
volumes_of_particles = PALSE_DS_manipulator.convert_listoflists_to_list(volumes_of_particles)
residue_volumes.append(sum(volumes_of_particles))
PALSE_statistic_handle.hist2d(residue_volumes, "residue_volumes_histogram.ps", 'Volume_of_Residues', 16, 20)
print("Histogram stored in residue_volumes_histogram.ps")
distribution_residue_volumes()
