Template C++ / Python API for developping structural bioinformatics applications.
User Manual
Buried_surface_area
Authors:F. Cazals and T. Dreyfus
Introduction
BSA. Consider a complex involving two species, and , called partner's labels — see the package MolecularSystemLabelsTraits for a description of such labels. Denote and their associated molecular models. In general, each such model is represented by a collection of balls defining the partner's solvent accessible model. With these notations, the complex involving the two partners is defined by .
The Buried Surface Area (BSA for short) of the molecular model is the area of the surface of and which gets buried within .
More formally, let be the boundary of the union of balls in , and likewise for . The BSA is defined by the surface area exposed by the individual partners, minus that of the complex:
.
The BSA can be assigned to the individual partners, which yields an algorithms to compute it. To see how, let be the surface of restricted to the exposed surface of , that is, prosaically, the contribution of to the exposed surface of the complex.
The BSA associated with the partners verifies
,
so that one has
.
This package. Provides tools to
compute the BSA of one partner in a molecular structure, possibly at different scales (the label of the partner may be primitive or hierarchical);
possibly serialize the BSA on a per particle basis.
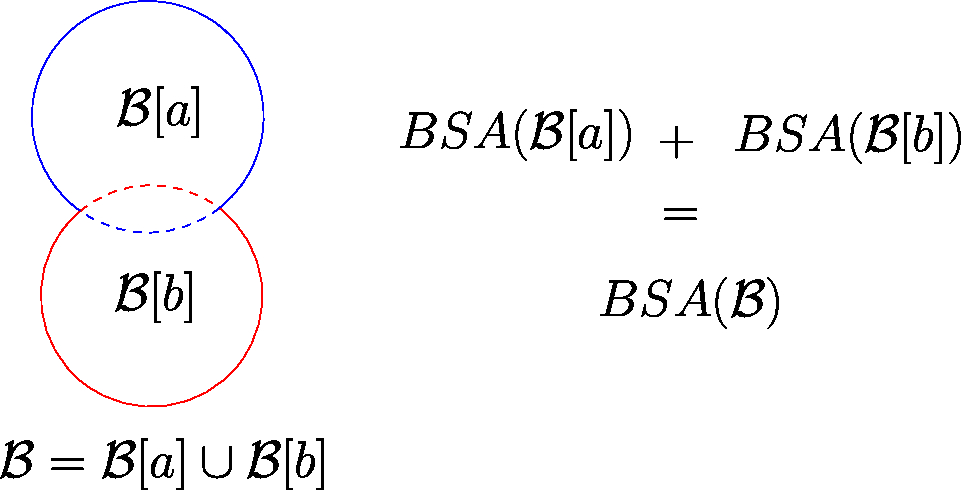
Illustration of BSA computation of a binary complex The molecular complex is decomposed using two labels (blue) and (red). The straight circular arcs are the surfaces of the restrictions and , while the dashed circular arcs are the buried surfaces of and . The BSA of is the sum of the BSAs of each restriction.
Algorithms
In order to compute the BSA in the context of hierarchical labels (see the package MolecularSystemLabelsTraits for a description of this hierarchy), we need to extend the formulae in the section Introduction .
Remind that in this context, labels are the nodes of rooted trees. Let be a hierarchical label such that it is a root of one of these trees, and the set of primitive labels having as ancestor in the tree. The formulae in the section Introduction apply for but not for labels in , since for some couple of labels in , the associated restrictions possibly intersect : this means that we cannot sum the BSA of the restrictions to the labels in for computing the BSA of .
To overcome this difficulty, we consider only the restrictions to a label that is a root of a tree. Thus, there are two cases for computing the BSA associated to a label :
is a root of a tree and we can apply formulae in section Introduction .
is not a root of a tree: considering the root of the tree containing , we use the restriction to for computing the BSA, but we only count the area of the surface of balls having the label ;
In this way, the BSA of any label remains additive. This case is illustrated on Fig. fig-bsa-example-IGAg with a IG-Ag complex.
There are two important remarks related to the computation of the BSA for any restriction of a molecular structure:
the BSA depends on the mediators: if a partner's particle with a label is partially buried in an interfacial mediator's particle, the buried portion of this partner's particle counts in the computation of the BSA. However, non-interfacial mediator's particles does not count in the computation. To distinguish these two types of mediator's particles, we just consider the non-interfacial mediator's particles as part of .
the BSA does not depend on the extras: as for the non-interfacial mediator's particles, the extra particles does not count in the computation of the BSA, and we treat them in the same way.
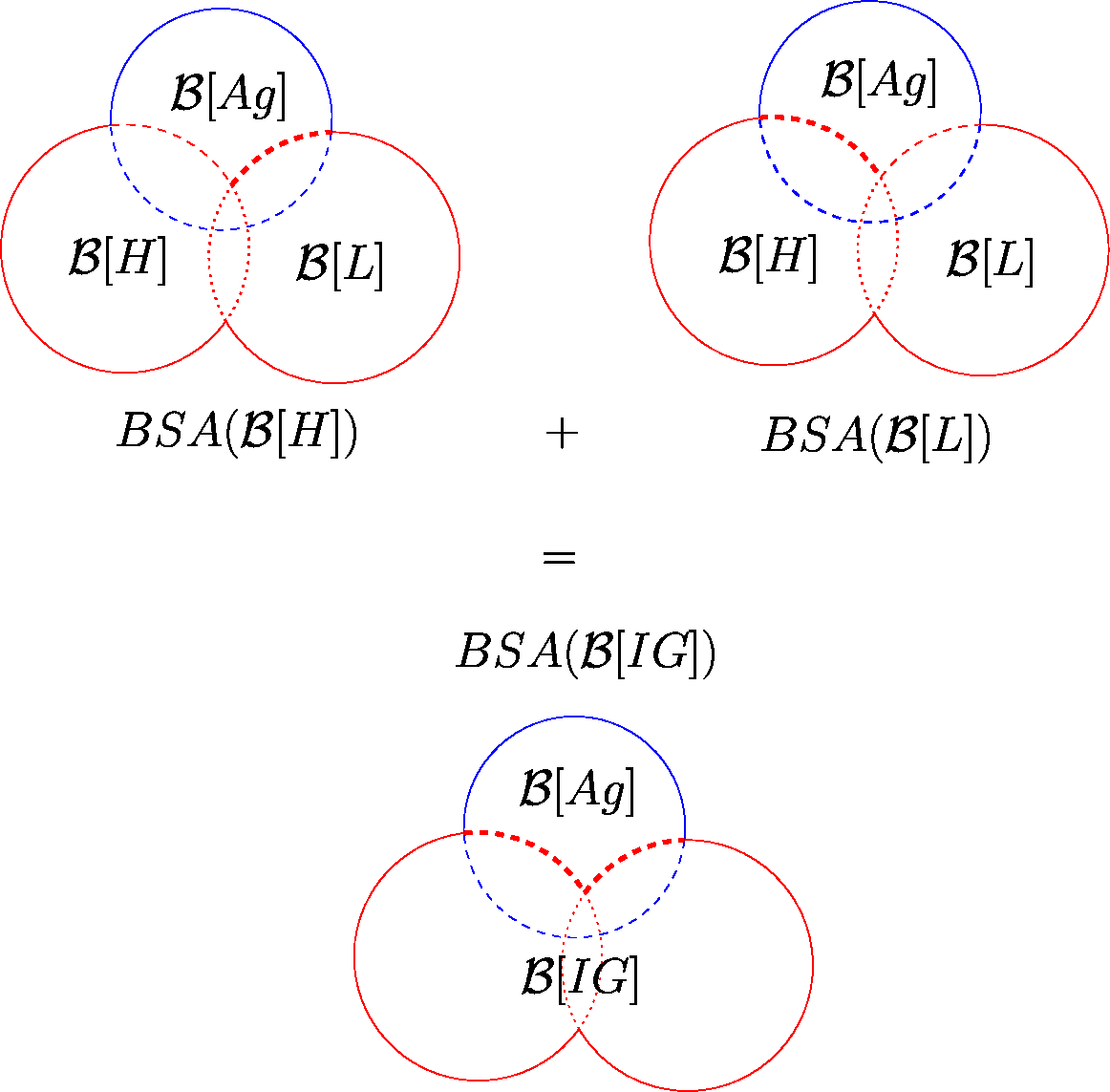
Illustration of BSA computation of a IGAg complex The immunoglobulin (IG) in red is decomposed onto its heavy (H) and light (L) chains. The antigen (Ag) is in blue. The straight circular arcs represent the exposed surface of the whole complex, and the non straight circular arcs represent the buried surface of each restriction in the whole complex. The dashed circular arcs represent the surface of each restriction that is buried in the whole complex, but exposed in its own partner. The sum of the BSA of the restrictions to H and L is the BSA of the restriction to IG.
Implementation
In the context of a molecular structure, each particle has a system's label identifying which partner, mediator or extra partner it belongs to. This information is mandatory for computing the BSA, but it may happen that in some context, the partners are not clearly identified, so that the particles has no label.
In section Main classes, one can find the two main classes provided by the package Buried_surface_area corresponding to the two possible situations. Then, each of these two main class leads to a module shown in section Modules .
With labels. When the partners can be specified using labels, one should use the class SBL::CSB::T_Buried_surface_area_with_labels< ParticleWithSystemLabelTraits , ParticleBSARecord , FT > : it is a functor (1) computing the BSA associated to a partner's label using the algorithm described in section Algorithms , and (2) storing the BSA on a per particle basis, using a type Particle_BSA_record – see below.
The three template parameters read as follows:
The first template parameter is a traits class defining a particle with an attached system's label, as defined in the package ParticleTraits.
The second template parameter is a class used to record BSA information on a per particle basis. In addition to the BSA, when the particle is an atom, one record the atomid, the resid, the chain id, etc; when the particle is a pseudo-atom, one may want to record the coordinates, radius, etc. Default implementations of these behaviors are provided with the classes SBL::CSB::T_BSA_record_atomic and and SBL::CSB::T_BSA_record_pseudoatom . Nb: the type corresponding to this second template parameter is passed to the BSA algorithm as a type of the traits calss passed to the class SBL::Modules::T_Buried_surface_area_with_labels_module< ModuleTraits > , see below.
The third template parameter is the type of the returned BSA (it is set by default to an interval certified to contain the true surface area). See Computing the BSA with partner's labels for an example.
Without labels.
when the partners cannot be specified using labels (e.g there is no label), one should use the class SBL::CSB::T_Buried_surface_area_without_label< ParticleTraits , FT > : it is a functor for computing the BSA associated to a given partner. In this version, no partner label is attached to the particles, but the functor takes as argument a predicate checking that a particle is in the partner for which we want the BSA. See Computing the BSA with a predicate for an example.
While the first version takes advantage of the knowledge on the composition of each partner for computing the BSAs of all primitive partners at the construction of the functor, the second version recompute the volume of the input partner each time the functor is called. Furthermore, there is no notion of hierarchy in the second version.
by providing the alpha-complex of the molecular structure.
by providing a range of particles defining the molecular structure (in this case, it is possible to add a constant value (i) to the radius of all particles and (ii) to the squared radius of all particles. For a discussion about these two parameters, see the section ...).
Once the object is constructed, it can be used as a function taking as argument:
In this example, atoms of a molecule are loaded from a PDB file, then annotated with default radii and systems' label, and inserted in a -complex data structure. Then, the BSA data structure is created from the -complex and the BSA of each chain is then printed.
Using the module computing the BSA with partner's labels
This example does the same work as the example of section Computing the BSA with partner's labels, but using the module SBL::CSB::T_Buried_surface_area_with_labels_module.
This example shows how to compute the BSA associated to selected partners in a molecular structure when atoms do not have an associated system's label. Assuming the input molecule is composed of three chains A, B and C, three predicates are defined for checking whether an atom is in a chain or another one. Then, after loading, annotating the particles and inserting them in the -complex, the BSA is computed for each chain using the three predicates.
Compute the buried surface area of two given partners.
Definition Buried_surface_area.hpp:228
Using the module computing the BSA with a predicate
This example generalizes the work done by the example of section Computing the BSA with a predicate by using the module SBL::CSB::T_Buried_surface_area_without_label_module. The main difference is that a generic predicate Is_in_partner is defined such that it is possible to iterate over the chains of the input molecule and to set the chain identifier of the current chain as the chain identifier of the predicate.


![${\cal B}[a_i]$](form_43_dark.png)
![${\cal B}[a_j]$](form_44_dark.png)
![${\cal B} = {\cal B}[a_i]\cup {\cal
B}[a_j]$](form_45_dark.png)

![$\partial{\cal B}[a_i]$](form_47_dark.png)
![${\cal
B}[a_j]$](form_48_dark.png)
![$BSA({\cal B}) = \marea{\partial{\cal B}[a_i]} + \marea{\partial{\cal B}[a_j]} - \marea{\partial{\cal B}}$](form_49_dark.png)
![$\partial{\cal
B}[a_i]\cap\partial{\cal B}$](form_50_dark.png)
![$BSA({\cal B}[a_i]) = \marea{\partial{\cal B}[a_i]} - \marea{\partial{\cal B}[a_i]\cap\partial{\cal B}}$](form_51_dark.png)
![$BSA({\cal B}) = BSA({\cal B}[a_i]) + BSA({\cal B}[b_i])$](form_52_dark.png)




![${\cal B}[l]$](form_60_dark.png)


![${\cal B}[a]$](form_55_dark.png)
![${\cal B}[b]$](form_56_dark.png)