Template C++ / Python API for developping structural bioinformatics applications.
User Manual
Protein_representation
Authors:F. Cazals and G. Carrière and T. Dreyfus and T. O'Donnell and R. Tetley
Illustration of Proteus, a god of changing nature, by Andrea Alciato. Similarly to Proteus, proteins keep changing shape, which is of paramount importance for the regulation of their functions.
Introduction
Biomolecules as linear polymers. Polypeptide chains (PCs) and nucleic acids are polymers of building blocks, namely amino acids and bases respectively. Both come with with topological, geometric, and biophysical information. These two biomolecules are therefore examples of linear polymers, represented using the package Linear_polymer_representation.
This package focuses on protein-specific functionalities; the reader is referred to the package Linear_polymer_representation for generic properties.
Functionalities offered. Because of these varying needs, this package provides functionalities to access all the information available at once. The main classes provided are:
Class SBL::IO::T_Protein_representation_loader : to create instances of the previous two classes. Recall that a PDB file may define several geometric models, typically if the PDB file comes from NMR. If so, the loader forces the choice of one model. For this model, a SBL::CSB::T_Protein_representation containing instances of SBl::CSB::T_Polypeptide_chain_representation is returned.
Instantiations on the shelf. We have discussed in the package Molecular_covalent_structure the hierarchy and variety of molecular covalent structures. This variety is mirrored by the following instantiations of protein representations – defined in Protein_representation_instantiations.hpp:
Note also that if there are missing residues in the input PDB file, the chain will be constructed with the missing residues. As a result, the corresponding SBL::CSB::T_Polypeptide_chain_representation will have several connected components. See the package Molecular_covalent_structure for further details.
In the sequel, we show how to access the various pieces of information. The following point should be stressed:
SBL::IO::T_Protein_representation_loader::get_protein (0) : returns the SBL::CSB::T_Protein_representation associated with the first PDB file loaded (if the loader was fed with several PDB files).
//Loads the covalent structure and the conformation from a PDB file.
for(Polypeptide_chains_iterator it = prot.chains_begin(); it != prot.chains_end(); ++it){
Polypeptide_chain_representation& P = it->second;
Covalent Structure File Loader statistics:
Number of loaded covalent structures: 1
Details for each covalent structure :
-- structure 1:
-- -- Number of loaded atoms: 3408
-- -- Number of particles: 5406
-- -- Number of modeled particles: 3408
-- -- Number of loaded conformations: 1
-- -- Number of bonds: 5467
-- -- Number of modeled bonds: 3469
-- -- Number of built disulfide bonds: 4 / 4
A common difficulty with PDB files is the presence of non-standard molecules / residues. Since this package is concerned with protein chains only, upon facing a residue which is non standard for a protein, the loader issues a message and halts the construction of the molecule. In other words, for a file containing a mixture of molecules (proteins, nucleic acids, drugs, etc), the loader only returns the protein chains found.
Enumerating atoms, residues, and the associated information
In the sequel, we focus on SBL::CSB::T_Polypeptide_chain_representation , and show how to access information associated with atoms. As an illustration, the first part of the snippet shows how to count elements of each type via a map; the second one collects the temperature factors of all atoms.
In terms of data structures, dereferencing the iterator on atoms via (*it) gives access to the Molecular_system::Molecular_atom data structure.
//Count the number of elements of each type :
//Compute min / max / average of temperature factors :
double min_B = 0, max_B = 0, ave_B = 0;
std::cout << std::endl << "Number of elements of each type : " << std::endl;
std::map<std::string, std::size_t> elements;
for(Polypeptide_chain_representation::Atoms_iterator it = P.atoms_begin(); it != P.atoms_end(); ++it)
std::cout << std::endl << "min / max / average of temperature factors : " << min_B << " " << max_B << " " << ave_B << std::endl;
Number of elements of each type :
C : 613
H : 264
N : 193
O : 185
S : 10
min / max / average of temperature factors : 0 84.09 28.3898
In a number of protein chains, residues are accompanied by insertion codes. Resid and insertion codes are accessed directly from the residue as follows :
If one or more residues are missing in a chain, that chain contains several connected components. Several methods apply both to the whole chain, and to connected components. These methods are parameterized by an integer: by default, this number is negative and the method applied to the whole chain irrespective of connected components. If there are say n connected components, the index in 0..n-1 indicates which c.c. should be processed.
Iterating on the backbone
In the following, an iterator on the backbone is used to store all backbone atoms into three containers, respectively for Calpha, C, N. As previously, backbone atoms are returned as Molecular_system::Molecular_atom .
std::cout << std::endl << "Found in the backbone : " << cas.size() << " CA, " << cs.size() << " C, " << ns.size() << " N." << std::endl;
Found in the backbone : 129 CA, 129 C, 129 N.
We only iterate on those atoms which are provided in the PDB. That is, for a structure presenting gaps, backbone atoms in the gaps are skipped. Since the primary sequence information is easily accessed, users can identify gaps based on resids. It is also possible to iterate over the backbone for each connected component of the chain : the begin and end iterators are parameterized by the number of the connected component (if none is specified, the whole chain is considered). See the code snippet below for an illustration.
//Collect the CA, C and N in the backbone
for(std::size_t i = 0; i < P.get_number_of_components(); i++)
{
cas.clear();cs.clear();ns.clear();
for(Polypeptide_chain_representation::Backbone_iterator it = P.backbone_begin(i); it != P.backbone_end(i); ++it)
Number of residues of each type :
ALA : 12
ARG : 11
ASN : 14
ASP : 7
CYS : 8
GLN : 3
GLU : 2
GLY : 12
HIS : 1
ILE : 6
LEU : 8
LYS : 6
MET : 2
PHE : 3
PRO : 2
SER : 10
THR : 7
TRP : 6
TYR : 3
VAL : 6
Accessing the Cartesian atomic coordinates
In the following, we show how to compute the center of mass of Calpha carbons, in a pedestrian way.
The example also calls the function SBL::CSB::T_Polypeptide_chain_representation::compute_heavy_atoms_center_of_mass() , whose name is self-explanatory.
//Compute the center of mass of the CA
double x = 0, y = 0, z = 0, num_calphas = 0;
for(Polypeptide_chain_representation::Calpha_iterator it = P.calphas_begin(); it != P.calphas_end(); ++it)
{
x += P.get_x(*it); y += P.get_y(*it); z += P.get_z(*it);
num_calphas++;
}
x /= num_calphas; y /= num_calphas; z /= num_calphas;
Polypeptide_chain_representation::Point_3 com = P.compute_heavy_atoms_center_of_mass();
std::cout << std::endl << "Center of mass of CA : (" << x << ", " << y << ", " << z << ")" << std::endl;
std::cout << std::endl << "Center of mass of Heavy atoms: (" << com.x() << ", " << com.y() << ", " << com.z() << ")" << std::endl;
Center of mass of CA : (53.2452, -17.1442, 7.57498)
Center of mass of Heavy atoms: (53.1526, -17.1834, 7.31916)
(Advanced) Note that the functions get_x() get_y() get_z() retrieve cartesian coordinates from the conformation. As mentioned in Introduction, the Cartesian coordinates are available via two channels: the data structures for the information contained in the PDB / mmCIF file and the data structure for the conformation. Both are coherent if the coordinates are not changed; nevertheless, functions get_x() get_y() get_z() use the conformation.
Changing the Cartesian atomic coordinates
The following example shows how to change Cartesian coordinates:
//Translate the protein two angstroms in the x direction
for(Polypeptide_chain_representation::Atoms_iterator it = P.atoms_begin(); it != P.atoms_end(); ++it)
std::cout << "Distance between new and old center of mass: " << distance << std::endl;
Distance between new and old center of mass: 2
(Advanced) Following the remark above, upon changing the coordinates, Cartesian coordinates must be accessed via the conformation and not the Molecular_system data structures. This is naturally taken care of by the get_x() get_y() get_z() functions. (NB: to put it sharply, the user should not mine the data structures to access the hidden Molecular_system data structures!)
Accessing internal coordinates
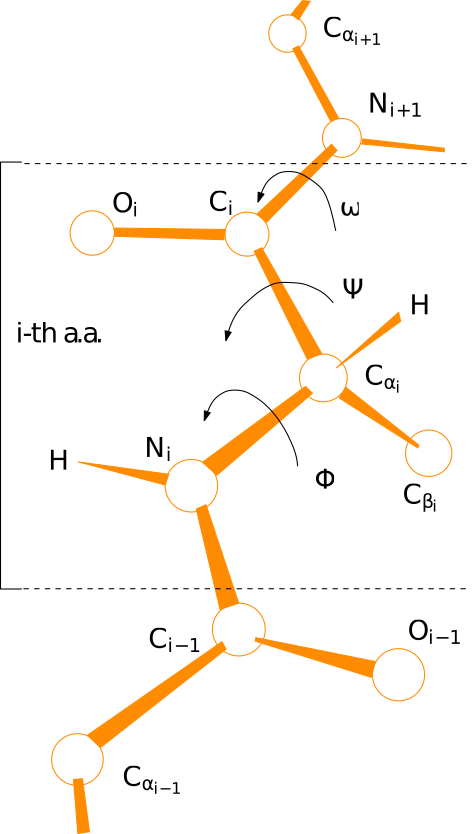
In the following, we show how to access internal coordinates, namely bond lengths, valence angles, and dihedral angles. Note in particular that the latter can be used to produce the so-called ramachandran plot. See also Fig. dihedral-angles-backbone.
This first snippet illustrates iterators returning all internal coordinates, which are accessed via dedicated iterators:
//Compute internal coordinates :
double mean_length = 0, num_bonds = 0;
for(Polypeptide_chain_representation::Bond_iterator it = P.bonds_begin(); it != P.bonds_end(); ++it)
{
mean_length += (*it).get_bond_length();
num_bonds++;
}
std::cout << "Mean bond length: " << (mean_length/num_bonds) << std::endl;
double mean_valence_angle = 0, num_angles = 0;
for(Polypeptide_chain_representation::Bond_angle_iterator it = P.bond_angles_begin(); it != P.bond_angles_end(); ++it)
Mean bond length: 2.0449
Mean valence angle: 1.63797
Mean dihedral angle: 0.222753
Mean phi angle: -1.05698
Mean psi angle: 0.52554
Mean omega angle: 0.425752
This second snippet focuses on the dihedral angles associated with a given residue:
Residue 2 has Phi angle: -1.87983, Psi angle: 2.15193 and Omega angle: 3.11996
Residue 33 has Phi angle: -1.04222, Psi angle: -0.895227 and Omega angle: -3.08817
Dihedral angles along the backbone of a polypeptide chain. By convention, the three angles display, namely , are associated with the i-th amino-acid.
Pairwise contacts
Various types of contacts are of interest in a polypeptide chain:
CT_COV: covalent contacts
CT_NCOV: non covalent contacts
CT_HBOND: contact corresponding to hydrogen bonds
CT_SB: salt bridges
CT_SS: disulfide bonds
The class SBL::CSB::Pairwise_contacts_for_polypeptide_chain contains a set of contacts for each of the previous types. By defaults, pairs of residues in contact, identified by their sequence number (i.e. resid) are recorded.
geometric information: Molecular_conformation, namely any data structure accommodating a conformation i.e. a d-dimensional point. The simplest such data structure is a vector of doubles, but more involved options are made available in package Molecular_conformation .
biophysical annotations: in Molecular_system, recall that a molecular system refers to a collection of molecular models – as found in a typical PDB file. Biophysical annotations, including the hierarchy associated with a PC (chain, a.a., atoms), are stored within
Molecular_system::Molecular_chain
Molecular_system::Molecular_residue
Molecular_system::Molecular_atom
The particular case of torsion angles
check here
Backbone torsion angles of a residue. The case of backbone torsion angles, that is for proteins is handled using the generic algorithm explained in section Torsion angles: generic algorithms – with a proper boundary check for the N-ter and C-ter residues.
Backbone torsion angles: iterators. These are boost filter iterators using a predicate has_type() indicating whether the instance's type matches that if the in the aforementioned Dihedral_angles_map_type.
For a angle for instance:
template <class ParticleTraits, class MolecularCovalentStructure, class ConformationType>
class T_Polypeptide_chain_representation<ParticleTraits, MolecularCovalentStructure, ConformationType>::Is_Phi_angle
{
public:
bool operator()(const Dihedral_angle& t)const{ return t.has_type(P_phi);}
};
typedef boost::filter_iterator<Is_Phi_angle, Dihedral_angle_iterator> Phi_iterator;
Side chain angles: iterators.check For angles of side-chains, we use the class SBL::CSB::Ordered_chain_iterator provided by the base class SBL::CSB::T_Linear_polymer_representation. The following dictionary is used to form the four-tuples of atom names used to infer torsion angles of side chains:
The class SBL::CSB::Chi_iterator is initialized with the N-CA-CB-x atom with x depending on the residue type. When incrementing, the next incident atom in the sidechain is found and the atoms in the Dihedral angle are consequently swapped. Knowledge on which atoms should be on the sidechain depending on the a.a type is stored in the SIDE_CHAIN_ATOMS_REFERENCE_TABLE static map. End on the sidechain is detected by keeping a count of the number of chi angles with an integer. For ALA and GLY, iterator end is set to begin and the dihedral angle attribute is set to null.
For proteins, the SIDE_CHAIN_ATOMS_REFERENCE_TABLE is as follows:
The loader SBL::IO::T_Protein_representation_loader has a number of options listed below, that can be set either from the command line using the Module_base framework, or directly using appropriate methods :
–coarse-levelint: coarse level for the covalent structure (0 : none, 1 : heavy atoms, 2 : residues)
(SBL::IO::T_Protein_representation_loader::set_coarse_level)
–no-ss-bonds: does not perform disulfide bonds calculations
(SBL::IO::T_Protein_representation_loader::set_without_disulfide_bonds)
–occupancy-policy: Selection policy for atoms with multiple alternate positions. Allowed values are: 1 (all), 2 (none), 3 (max, default), 4 (min). Atoms with only one position are always kept.
(SBL::IO::T_Protein_representation_loader::set_occupancy_factor)
–alternatestring: Alternate coordinates: alternative to be used (char)
(SBL::IO::T_Protein_representation_loader::set_alternate_coordinate)
–model-numberint: ID of the model to be loaded frome the PDB file (default is 1)
(SBL::IO::T_Protein_representation_loader::set_PDB_model_number)
–chainsstring: Subset of chains to be loaded.
(SBL::IO::T_Protein_representation_loader::set_loaded_chains)
–allow-incomplete-chains: Allows to load residues even if there are missing ones.
(SBL::IO::T_Protein_representation_loader::set_allow_incomplete_chains)
Particle info for proteins. The class SBL::CSB::T_Particle_info_for_proteins is a record providing the required information, namely: pointer to the atom itself, res_id, ins_code, res_name, atom_name, chain_id.
The particle info is used as a key in a map as follows:
typename T_Molecular_covalent_structure<ParticleInfo>::Particle_rep
T_Molecular_covalent_structure<ParticleInfo>::add_particle(const Particle_info& info)
{
Particle_rep p = boost::add_vertex(this->m_graph);
this->m_graph[p] = info;
// Particle_info to Particle_rep
this->m_particleInfo_to_particleRep.insert(std::make_pair(this->m_graph[p], p));
// ParticleRep to linearPosition
this->m_particleRep_to_linearPosition.push_back(p);
return p;
}
Molecular_covalent_structure. The covalent structure is represented using the class SBL::CSB::T_Molecular_covalent_structure. It is effectively built by the class SBL::CSB::T_Molecular_covalent_structure_builder_for_proteins, which creates the individual amino acids and links them into the graph representing the molecule. This creation involves all atoms of all residues, be they present in the PDB file or not – see the notion of embedded atom below.
In the following, we use the following types, as defined above:
Particle_info : information record on the atom.
Particle_rep : index of the corresponding vertex in the boost graph.
ParticleInfo_to_particleRep_map_type: Particle_info to Particle_rep.
Key members of the class regarding indices:
// The boost graph used to represent the structure
Covalent_structure_graph m_graph;
// Mapping particleInfo to graph vertex/particleRep
ParticleInfo_to_particleRep_map_type m_particleInfo_to_particleRep;
// Mapping the particleRep (an int, actually), to the linear position used in the conformation
std::vector<int> m_particleRep_to_linearPosition;
Note that the position is used in the conformation, not in the molecular covalent structure itself.
================where is this used??????????
(GC) is_residue_heavy_atoms_embedded works by counting the number of heavy atoms using Is_Heavy predicate and Residue_atoms_iterator, and checking the value depending on the residue type. Information on the number of heavy atoms per a.a type is stored in the RESIDUE_HEAVY_ATOM_SIZE_REFERENCE_TABLE static map.
Conformations. The class SBL::Model::T_Conformation_traits proposes to store a conformation as a vector of float types representing coordinates.
SBL::IO::T_Molecular_covalent_structure_loader_from_PDB : loads and delivers Covalent_structures
SBL::Models::T_PDB_file_loader loads a PDB \ mmCIF file (its models to be precise), and store the atomic information into an Molecular_system data structure.
T_Polypeptide_chain_representation. The high level class SBL::CSB::T_Polypeptide_chain_representation proposes high level operations, in particular the ability to obtain internal coordinates from atomids – specified in the PDB file. Such operations use the internal representation of atoms via their particle representations. As an example, consider:
To provide these high level operations, we use the map:
//! The covalent structure of the protein
Molecular_covalent_structure& m_covalent_structure;
//! The conformation of the protein
Conformation_type m_conformation;
// mapping a \sbl_ref_package{Molecular_system} atom via its atom id to the corresponding Particle_rep ie Particle_vertex_descriptor
typedef typename std::map<unsigned, Particle_rep> AtomId_to_particleRep_map_type;
AtomId_to_particleRep_map_type m_atomId_to_particleRep;
//! Return the value of this angle.
FT get_valence_angle() const
{
//get the internal particle reps
Particle_rep a = this->m_P->m_atomId_to_particleRep.at(boost::get<0>(this->m_bond_angle).atom_serial_number());
Particle_rep b = this->m_P->m_atomId_to_particleRep.at(boost::get<1>(this->m_bond_angle).atom_serial_number());
Particle_rep c = this->m_P->m_atomId_to_particleRep.at(boost::get<2>(this->m_bond_angle).atom_serial_number());
//get the interal bond reps
std::pair<bool, Bond_rep> bond1 = this->m_P->m_covalent_structure.get_bond_rep(a, b);
std::pair<bool, Bond_rep> bond2 = this->m_P->m_covalent_structure.get_bond_rep(b, c);
assert(bond1.first && bond2.first);
//get the internal bond angle rep
std::pair<bool, Bond_angle_rep> bond_angle = this->m_P->m_covalent_structure.get_bond_angle_rep(bond1.second, bond2.second);
//return the valence angle
return this->m_ic.get_valence_angle(this->m_P->m_conformation, this->m_P->m_covalent_structure, bond_angle.second);
}
Coherence. For polypeptide chains represented by the class SBL::CSB::T_Protein_representation, the coherence between these maps is ensured by the following function
void T_Polypeptide_chain_representation<ParticleTraits, MolecularCovalentStructure, ConformationType>::
build_model_to_covalent_structure_mapping()
{
for(Particles_iterator it = this->particles_begin(); it != this->particles_end(); it++){
this->m_atomId_to_particleRep[(*this)[*it]->atom_serial_number()] = *it;
this->m_covalent_structure.set_particle_linearPosition((*it),m_atomId_to_particleRep.size()-1);
}
};
Due to the previous, the iterators of m_covalent_structure can now be used to extract particles and compute internal coordinate values using m_conformation as the coordinates of particles or for any other purpose requiring simultaneous use of m_covalent_structure and m_conformation.
Applications
This package also offers several useful programs to inspect properties of proteins / their conformations:
sbl-protein-info.exe: loads protein chain(s) form a PDB file, and parses it to deliver a variety of statistics (number of a.a. of each type, center of mass of the structure, calculation of internal coordinates and average statistics, etc.) Note that the corresponding code is of interest for developers wishing to use this package to develop novel applications.
sbl-protein-ramachandran.exe: computes the Ramachandran plot of a protein; alternatively, for conformations of the same protein, computes the Ramachandran plot of specified amino-acids.
sbl-protein-pdb-cleaner.py : a python script cleaning PDB files to make sure the contain the required information for loaders (in particular chemical element names), and builders of the covalent structure (correct atom naming required to build the molecular topology).