Template C++ / Python API for developping structural bioinformatics applications.
User Manual
MolecularSystemLabelsTraits
Authors:F. Cazals and T. Dreyfus
Introduction
This package defines a C++ concept to perform a hierarchical decomposition of a molecular structure based on labels. The primary goal of such decompositions is to study interfaces between groups of atoms/particles–see the application Space_filling_model_interface.
We assume a list of labels meant to tag the particles, defined as the union of three lists:
the list of partners,
the list of mediators,
the list of extra partners.
The semantics associated with these lists of labels is the following:
the partners' labels: these labels identify all the particles involved in molecular interfaces of interest.
the mediators' labels: these labels identify all the particles sandwiched in-between two particles whose labels are partners' labels.
the extra partners' labels (extra for short): these labels identify all the remaining particles (ions, metabolites, etc).
We assume that the labels in each of these three lists correspond to the vertices of rooted trees in a forest. The leaves of these trees are called primitive labels, and the internal vertices are called hierarchical labels. The set of all primitive labels is denoted , and given a label , the set of primitive labels in the sub-tree rooted at is denoted . Note that if is a primitive label, then .
Given the list of particles of a molecular structure , the primitive labels induce a partition of . The restriction of to the label consists of the set of particles within having as label.
Note that abusing terminology, we may use partner and mediator to refer either to the labels or the systems associated to these labels.
Since labels are assumed to have a hierarchical structures in terms of rooted trees, the forest associated to the partners is called the partners' forest, and the one associated to the mediators is called the mediators' forest.
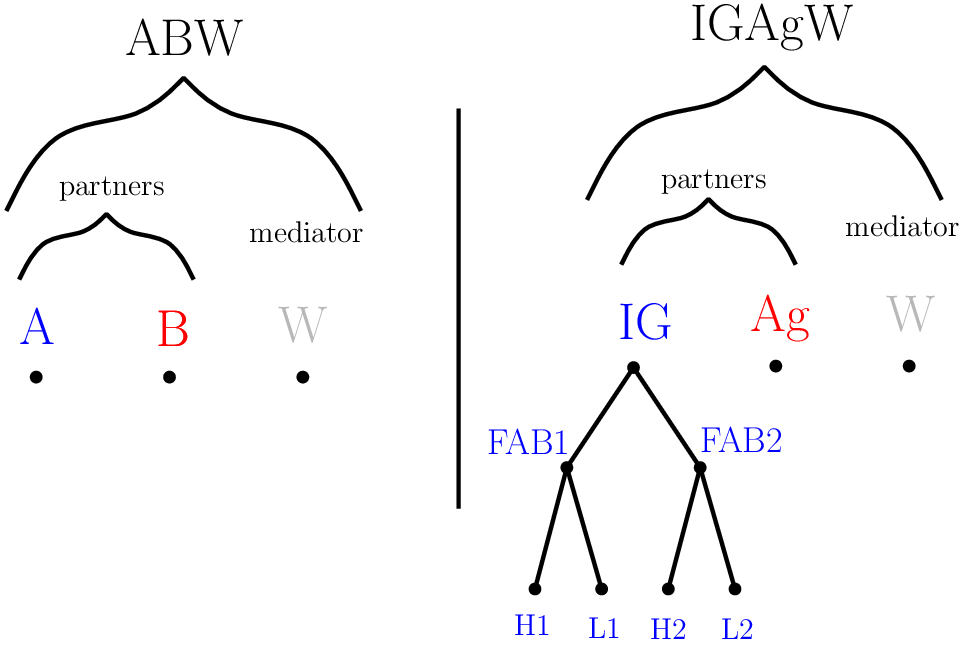
The simplest case corresponds to a protein-protein complex, with the partners' forest reduced to two trees, each reduced to its root, which defines a primitive label – see Fig. fig-example-labels-forest (Left).
An antibody (or immunoglobulin or IG) is a molecule from the immune system coming in several isotypes, see the Wikipedia Antibody page. Consider an antibody whose isotype is IgG–the IG involves one monomer of two heavy and two light chains, and assume this IG makes up an IG - Ag complex. Such a complex can be modeled with two partners' labels, that of the receptor (the IG) and that of the ligand (the Ag). Moreover, the partner label for the IG can be modeled in a hierarchical fashion: the IG has two fragment antigen binding (FAB) regions, each involving one heavy and one light chain. This results in a partners' forest with five primitive labels (the Ag, the two heavy chains and the two light chains of IG), and three hierarchical labels (the two FAB regions and the IG itself) – see Fig. fig-example-labels-forest (Right).
Note that the decomposition can even be refined, since the variable domain of each chain can be decomposed into three complementarity determining regions (CDR) flanked by four framework regions.
Examples of Labels' Forest : ABW and IGAgW
Partners' and Mediators' labels forest in two cases: (Left) a binary complex (partners A and B) mediated by water molecules (W), and (Right) a immunoglobulin (IG)/ antigen (Ag) complex mediated by water molecules (W).
Using existing models in existing Applications : I/O
From the user standpoint, using programs resorting to models of MolecularSystemLabelsTraits has two implications:
(i) the models used by these programs represent different molecular systems. The simplest case is that of a binary complex, e.g. an antibody/antigen system. Thus, the calculations carried out and the corresponding output depend on the specification used – see example in section Example 1: Intervor .
(ii) the models may require different user specifications. For example, for a binary complex involving proteins, the user needs to specify which polypeptide chains define each partner. Such specification is typically passed through the command line of the program (see e.g. sections Example 1: Intervor). If on the other hand the specification is more complex, e.g. the decomposition of a polypeptide chain into user defined domains (domains for short in the sequel), a file may be used to pass it.
To ease things, when an application involves programs resorting to different models of MolecularSystemLabelsTraits, the name of each program contains a keyword identifying the type of partners and mediators (if any) (see section Example 1: Intervor).
When the name of the program contains the keyword "domain", the model is SBL::Models::Domain_labels_traits. This model requires the user to specify a file describing the decomposition of the input polypeptide chains into their domains. A complete description of this specification file can be found in the reference manual of SBL::Models::Domain_labels_traits (see section Example 2: Binary Interface Finder (BIF)).
Example 1: Intervor
The application Space_filling_model_interface provides methods to study interfaces between partners of a molecular structure. Different strategies to define these partners yield different programs using different models of MolecularSystemLabelsTraits :
: a program studying interfaces between two partners, called A and B for convenience, in the molecular structure. Defining which chains of the molecular structure define A or B is done using the command-line option –partners twice: the first occurrence lists the chains in A, the second occurrence lists the chains in B. The program produces an XML archive containing information and statistics on the different interfaces between A and B, including the mediated interfaces.
: a program studying interfaces in an antibody - antigen complex, the antibody consisting of two heavy and light chains. Defining which are the chains of the antigen, and the light and heavy chains of the antibody, is done using the command-line options –ag, –igL and –igH. The program produces an XML archive for each pair of partner's labels that have no common ancestor, each XML archive containing information and statistics on the different interfaces between the corresponding partner's labels, including the mediated interfaces.
Note that in the name of both programs, the letter means that the inter-facial water is used for mediating contacts between two partners.
Example 2: Binary Interface Finder (BIF)
The application Space_filling_model_interface_finder finds possible interfaces in a molecular structure, producing a graph with one vertex per primitive partner, and an edge connecting two primitive partners sharing an interface. Two programs use two different models of MolecularSystemLabelsTraits, so as to specify the notion of partner:
: each chain is a partner in the molecular structure. In this case, there is no command-line option, since all the information is contained in the input PDB file.
: the partners are defined by group of chains, each of them possibly decomposed by domains. In this case, the user has to specify how to group the chains, and how to decompose the desired chains into domains. This is done with a specification file passed with the command-line option –domain-labels, as described in the reference manual of the class SBL::Models::Domain_label_traits.
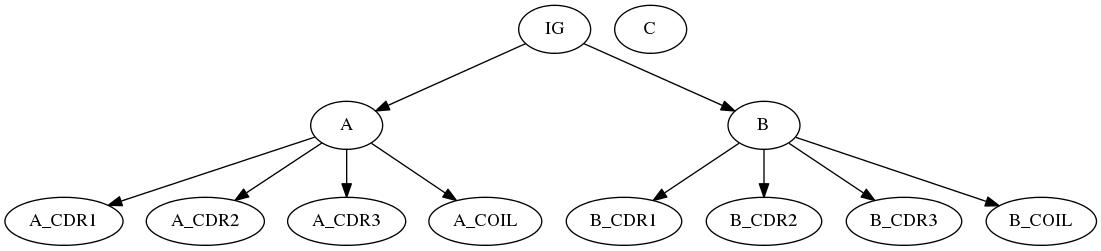
Here is an example of such a specification file for decomposing the immunoglobulin / antigen complex 1a2y (IMGT version) by complementarity determining regions (CDR):
(i) by creating a template of decomposition of chains (keyword domains-template-begin)
(ii) by enumerating the chains of the molecular structure, specifying if necessary the template they are following (keyword chains-enumeration-begin)
(iii) by grouping the chains hierarchically (keyword chains-hierarchy-begin)
#example with 1a2y decomposed by CDR
#the first line starts the template and give it a name
domains-template-begin CDRS
#the following lines contain: the name of the label, then the ranges
#of residues corresponding to this label (including the bounds)
CDR1 27-38
CDR2 56-65
CDR3 105-117
#the star denotes the complementary, i.e all residues not mentionned
#before in the template, the second star indicates that each connected
#component of the complementary will be attributed a different label
COIL **
#terminates the template
end
#enumerates the chains and possibly associates template to them
chains-enumeration-begin
AB like CDRS
C
end
#groups hierarchically the chains
chains-hierarchy-begin
IG A B
end
Labels and their complement aka coil regions
The previous example illustrates an interesting feature, as one specifies
regions – the CDR
the complement of these regions, with the directive COIL.
More generally, the COIL directive can be used in three ways:
no COIL directive: atoms which do not belong to any domain specified are grouped under a label named OTHER.
COIL : a single star symbol ("</em>") means that all the complementary regions of the specified regions are grouped under a unique label COIL.
COIL : two star symbols ("</strong>") are found, one label is created for each connected component. The name for each label is the number of the connected component (starting at 1).
Note also that there is always a created label for all residues in the complementary that are located after the residue with the largest id in the specification file, even if there is no such residue in the input PDB file.
Using existing models to develop novel applications
In this section, the existing models of MolecularSystemLabelsTraits are discussed. These models are divided into two groups: the models that are typically used for partners and those used for mediators. There is also one model for representing extra particles, namely SBL::Models::Extra_labels_traits, that provides one label "X" for all extra particles. We also discuss how to combine specifications for partner, mediators and extra partners.
SBL::Models::AB_label_traits: defines two partners from two groups of chains specified by the user, option –partner).
SBL::Models::IG_label_traits: defines a hierarchy of labels for an immunoglobulin, which is decomposed into heavy and light chains. The user must specify which chains are the light and heavy chains, with the options –igL and –igH.
SBL::Models::IGAg_label_traits: as above, except that a second partner corresponding to the antigen is specified. The specification of chains making up the antigen is done using the option –ag).
SBL::Models::Domain_label_traits: defines a hierarchy of labels using a decomposition of polypeptide chains into user defined domains, as specified in file loaded with the option –domain-labels. See example of section Example 2: Binary Interface Finder (BIF) for two decompositions, one at the chain level, the other one at the domain level.
returning the primitive label of a given particle.
However, when a molecule has partners, mediators and possibly extra partners, one needs a mechanism to load all user defined specifications and to check in which partner / mediator / extra partner a particle belongs to. The class SBL::IO::T_Primitive_labels_loader< PartnerLabelsTraits , MediatorLabelsTraits , ExtraLabelsTraits > is a model of the Loader concept loading the user defined specifications and initializing three instances of the class Primitive_label_classifier for partners, mediators and extra partners. Then, particles enriched with labels update their label with the method SBL::Models::T_Particle_with_system_label_traits::Particle_type::set_system_label, that uses the three classifiers of primitive labels.
Example: Dumping the Forest of Labels for Partners
Decomposing an immunoglobulin (IG) - antigen complex (PDBID: 1a2y). The IG consists in a light chain (A) and a heavy chain (B), whose variable domains are decomposed into complementarity determining regions (CDRs). The antigen consists in the chain C.
Label type: enumerates all the labels of the forest; in particular, it enumerates first the primitive labels, and then the hierarchical labels.
get_number_of_labels static method: returns the number of labels in the forest,
get_number_of_primitive_labels static method: returns the number of primitive labels in the forest,
get_parent_of static method: given a label of the forest, returns its parent if any, the label itself otherwise,
to_string static method: given a label of the forest, returns its string version.
the Primitive_label_classifier class: a functor that, given a particle, returns a pair (boolean, Label) stating if the input particle has one of the primitive label defined in this class. If true (i.e. the boolean holds True), the label returned is the primitive label. Note that the classification of particles may depend on user defined data (see the example of user defined protein domains above) : the Primitive_label_classifier class defines methods for loading the user-specified data (see examples in sections Example: One label and Example: Hierarchy of labels .
As discussed above, the C++ model SBL::Models::IG_label_traits provides a simple decomposition of an immunoglobulin onto its heavy and light chains.
Note that the identification of the H and L chains must be provided by the user. In the code below, the H and L chains are defined from the command line using the SBL::Models::IG_label_traits::Primitive_label_classifier defines the requirements of the Loader C++ concept, as explained in the package Module_base,
allowing to define command line options for defining which are the chains in the input PDB file corresponding to the light and heavy chains.









![$ {\cal B}[l]$](form_930_dark.png)