|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
![]()
Authors: F. Cazals and T.Dreyfus
Goal. Molecular distances aim to compare different conformations of the same molecule or complex.
A conformation is represented either in Cartesian coordinates (using the Point_d format) or in internal coordinates. Computing distances between conformations occurs in essentially two situations:
In the former case, it is clear that the same atoms are available. In the latter, the hydrogen atoms are typically not reported in a coherent way.
Modes. For distances based on Cartesian coordinates, this account for the following four modes used in distance calculations:
As noticed in Introduction, a mapping between atoms is required to compute distances based on Cartesian coordinates. The trivial identity mapping is computed as follows:
Chains. It is assumed that the two files compared contain the same number of chains – if applicable. The trivial mapping between chains is used – i-th chain of the first file compared with the i-th chain of the second file.
Amino-acids. When comparing two chains, a.a. acids are sorted by residue sequence number, and common a.a. are selected. (NB: this allow handling the case where one chain misses selected a.a., e.g. located in a flexible loop.)
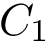 and
and 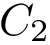 be two conformations of a molecule of
be two conformations of a molecule of 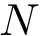 atoms. We note
atoms. We note  the
the 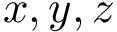 coordinates of the
coordinates of the 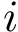 -th atom of the conformation . The rigid registration of over is denoted
-th atom of the conformation . The rigid registration of over is denoted 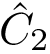 , and its coordinates
, and its coordinates 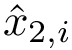 . The least-RMSD of and is the root mean square distance between coordinates of and
. The least-RMSD of and is the root mean square distance between coordinates of and 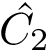 :
: 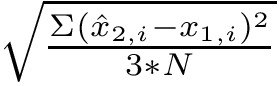
The rigid registration is performed using the package Point_cloud_rigid_registration_3. Note that this rigid registration takes only account for rotations and translations. In this package, we also need to take account for chirality, so that we need in addition a mirror transformation in the registration.
![$[0, 2\pi]$](form_756.png) . The angular distance is a RMSD where the pairwise distances between particles is defined over .
. The angular distance is a RMSD where the pairwise distances between particles is defined over .
The previous distances were RMSD defined for pairs of particles. Another way to compare conformation is to compute the RMSD between all possible pairs of particles of one conformation with all matching pairs of particles of the other conformation. The resulting distance is called the RMSD internal distance.
This package offers also the possibility to plug any executable that takes two input D dimensional points and returns a single value distance. In this way, any other kind of distance, that is externally defined, can be used in the SBL.
Except for the distance described in section Other distances, all described molecular distances require an alignment process to match particles of the two input conformations.
The concept MolecularAlignment is a simple functor taking as input the position of a particle in a conformation and a boolean tag indicating the direction of the alignment (first conformation to second, or opposite direction). It then returns the position of the input particle in the other conformation. The default functor is SBL::CSB::Molecular_alignment_default and simply returns the input position, assuming that both conformations are already aligned.
The l-RMSD and the RMSD internal distances require both a geometric definition of a particle. As mentionned, a particle is represented by a 3D point. Thus, the concept GetParticle defines a functor that takes as input a conformation and a position, and returning a 3D point corresponding to the target particle. The default functor is SBL::CSB::T_Get_particle_default and simply returns a CGAL Point_3 structure of the ith particle in the input conformation.
The l-RMSD is defined in the class SBL::CSB::T_Least_RMSD_cartesian< Conformation , GetParticle , MolecularAlignment > . The parameter Conformation is the representation of the input conformations and should be compliant with the CGAL::Point_d class of the CGAL library. The parameters GetParticle and MolecularAlignment were both previously described.
In addition, the class SBL::CSB::T_Least_RMSD_cartesian_with_chirality< Conformation , GetParticle , MolecularAlignment > computes the distances between mirror images of the input conformations, and returns the smallest distance.
The RMSD internal distance is defined in the class SBL::CSB::T_Squared_RMSD_internal_distance< Conformation , InternalDistance , GetParticle , MolecularAlignment > . The parameter InternalDistance is a functor for computing the distance between two particles in the same conformation. The three other parameters were all previously described. Note that this functor returns the squared distance, avoiding the use of the square root operation, that might not be necessary.
Externally defined distances can be wrapped using the class SBL::CSB::T_External_distance< Conformation , FT > . Parameters were all already described. The only additional requirements are :
The following example loads two conformations from two input files, and computes the least-RMSD between those two conformations.
The following example loads two conformations from two input files, and computes the RMSD internal distance between those two conformations.
The following example loads two conformations from two input files, and computes the distance from an external executable called sbl-conf-distance.exe.
This package also offers a variety of programs performing different tasks related to the least-RMSD:
See the following jupyter notebook:
The following sections provide examples for the following executables:
In the following, we compare two structure of the SARS-COV-2 receptor binding domain RBD.
In the options, note in particular:
In the output, the following is noticeable:
# A Small function launching the calculation for a pair of PDB files
from subprocess import Popen, PIPE
from subprocess import check_output, CalledProcessError, STDOUT
import subprocess
def cmp_structures(exe, pdb1, chains1, pdb2, chains2, grain_mode):
cmd = [exe, "-f", pdb1, "--load-chains", chains1, "-f", pdb2, "--load-chains", chains2, "--grain-mode", grain_mode, "-v", "--xyz"]
s=subprocess.check_output(cmd, encoding='UTF-8')
print(s)
cmp_structures("sbl-lrmsd-for-pdb-pair.exe", "data/6w41.pdb", "C", "data/6m0j.pdb", "E", "0")
cmp_structures("sbl-lrmsd-for-pdb-pair.exe", "data/6w41.pdb", "C", "data/6m0j.pdb", "E", "1")
cmp_structures("sbl-lrmsd-for-pdb-pair.exe", "data/6w41.pdb", "C", "data/6m0j.pdb", "E", "2")