|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
![]()
Authors: S. Marillet and P. Boudinot and F. Cazals
This package provides tools to (i) select binding affinity prediction models from atomic coordinates, typically crystal structures of the partners and/or of the complex, and to (ii) exploit such models to perform affinity predictions.
At the selection stage, given a set of predefined variables coding structural and biochemical properties of the partners and of the complex, the approach consists of building sparse regression models which are subsequently evaluated and ranked using repeated cross-validation [124] .
At the exploitation stage, the variables selected are used to build a predictive model on a training dataset, and predict the affinity on a test dataset.
More precisely, the approach runs through 3 steps:
Consider two species 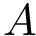 and
and 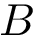 forming a complex
forming a complex 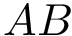 . The dissociation is defined by
. The dissociation is defined by ![$\Kd = [A][B]/[AB]$](form_41.png) , namely the ratio between the concentration of individual partners and bound partners.
, namely the ratio between the concentration of individual partners and bound partners.
The strength of this association is quantified by the dissociation free energy 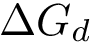 , which, in the
, which, in the 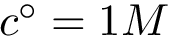 standard state, satisfies with
standard state, satisfies with 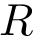 Boltzmann's constant and
Boltzmann's constant and 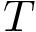 the temperature:
the temperature:
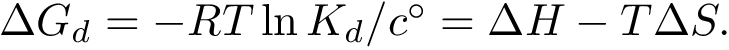
This equation shows that 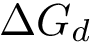 has two components respectively coding the enthalpic (
has two components respectively coding the enthalpic ( 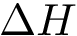 ), and entropic (
), and entropic ( 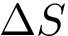 ) changes upon binding. As stated above, we wish to estimate these quantities from atomic coordinates, typically crystal structures of the partners and/or the complex.
) changes upon binding. As stated above, we wish to estimate these quantities from atomic coordinates, typically crystal structures of the partners and/or the complex.
Parameters
The parameters used in this package are based on the following key geometric constructions:
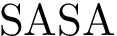 for short) is the sum of the surface areas exposed by the individual atoms. Upon complex formation, the buried surface area (BSA) is the surface area of the partners buried at the interface, namely the lost by the individual atoms. The solvent accessible surface areas are computed using
for short) is the sum of the surface areas exposed by the individual atoms. Upon complex formation, the buried surface area (BSA) is the surface area of the partners buried at the interface, namely the lost by the individual atoms. The solvent accessible surface areas are computed using 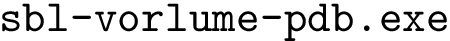 . See section Solvent Accessible Model for more details
. See section Solvent Accessible Model for more details 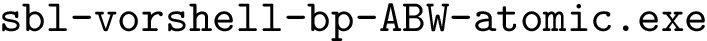 . See section Interfaces, Buried Surface Area, and Core-rim Models for more details.
. See section Interfaces, Buried Surface Area, and Core-rim Models for more details. 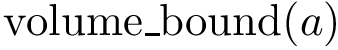 (resp.
(resp. 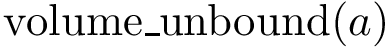 ) the volume of the Voronoi restriction of an atom
) the volume of the Voronoi restriction of an atom 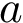 in the bound form (resp. unbound form). The volumes are computed using .
in the bound form (resp. unbound form). The volumes are computed using . 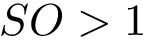 Thus, the SO generalizes core-rim models [100] , since the rim corresponds to
Thus, the SO generalizes core-rim models [100] , since the rim corresponds to 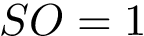 , and the core to . The shelling order of interface atoms is computed using . See section Interfaces, Buried Surface Area, and Core-rim Models for more details.
, and the core to . The shelling order of interface atoms is computed using . See section Interfaces, Buried Surface Area, and Core-rim Models for more details. Using the previous notions, we define four categories of atoms of a complex:
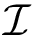 , we define two groups, those found on the rim (
, we define two groups, those found on the rim ( 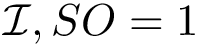 ), retaining solvent accessibility, and the remaining ones (
), retaining solvent accessibility, and the remaining ones ( 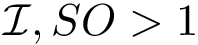 ).
). 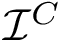 , we distinguish between those retaining solvent accessibility ( and
, we distinguish between those retaining solvent accessibility ( and 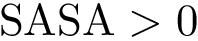 in the complex), and those which do not ( and
in the complex), and those which do not ( and 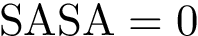 in the complex).
in the complex). Biophysical rationale.
Before defining our parameters, we raise several simple observations underlying their design:
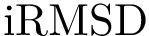 ). In the sequel, we refine this measure using atomic packing properties. Indeed, packing properties are intuitively related to the vibrational entropy of atoms.
). In the sequel, we refine this measure using atomic packing properties. Indeed, packing properties are intuitively related to the vibrational entropy of atoms. Using the quantities defined in section General pre-requisites, we define the following variables:
Interface terms.
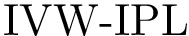 , see Eq. (eq-ivwipl): the sum over interface atoms of their shelling order normalized by their packing. Note that since a packed interface is more likely to result in a high affinity, the shelling order is weighted by the inverse of the volume, yielding the inverse volume-weighted internal path length .
, see Eq. (eq-ivwipl): the sum over interface atoms of their shelling order normalized by their packing. Note that since a packed interface is more likely to result in a high affinity, the shelling order is weighted by the inverse of the volume, yielding the inverse volume-weighted internal path length . 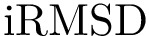 = Interface RMSD : the least RMSD for interface atoms.
= Interface RMSD : the least RMSD for interface atoms. Packing terms.
Consider the volume variation of 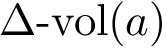 , see Eq. (eq-atom-volume-variation). As recalled above, packing properties are important in several respects. Adding up volume variations yields the following four sum of volumes differences (VD) parameters:
, see Eq. (eq-atom-volume-variation). As recalled above, packing properties are important in several respects. Adding up volume variations yields the following four sum of volumes differences (VD) parameters:
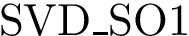 (
( 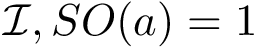 ; Eq. (eq-svdsoo)): sum of VD for the rim interface atoms.
; Eq. (eq-svdsoo)): sum of VD for the rim interface atoms. 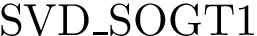 (
( 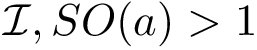 ; Eq. (eq-svdsogto)): sum of VD for interface atoms in the interface core.
; Eq. (eq-svdsogto)): sum of VD for interface atoms in the interface core. 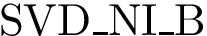 (
( 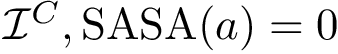 ; Eq. (eq-svdnibur)): sum of VD for buried non interface atoms.
; Eq. (eq-svdnibur)): sum of VD for buried non interface atoms. 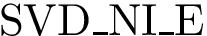 (
( 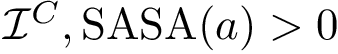 ; Eq. (eq-svdnisurf)): sum of VD for exposed non interface atoms.
; Eq. (eq-svdnisurf)): sum of VD for exposed non interface atoms. Solvent interactions and electrostatics.
The interaction between a protein molecule and water molecules is complex. In particular, the exposition to the solvent of non-polar groups hinders the ability of water molecules to engage into hydrogen bonding, yielding an entropic loss for such water molecules. We define the following terms:
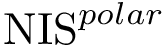 , see Eq. (eq-nispol): the fraction of charged a.a. on the non interacting surface, as defined in [108] .
, see Eq. (eq-nispol): the fraction of charged a.a. on the non interacting surface, as defined in [108] . 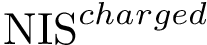 , see Eq. (eq-nischar): the fraction of polar a.a. on the non interacting surface [108] .
, see Eq. (eq-nischar): the fraction of polar a.a. on the non interacting surface [108] . 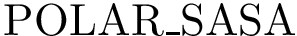 , see (Eq. (eq-polar_sasa): an intermediate-grained description of the non-interacting surface which consists in the atomic-wise polar area of the complex. The corresponding term, (Eq. (eq-polar_sasa)), is a weighed sum of exposed areas.
, see (Eq. (eq-polar_sasa): an intermediate-grained description of the non-interacting surface which consists in the atomic-wise polar area of the complex. The corresponding term, (Eq. (eq-polar_sasa)), is a weighed sum of exposed areas. 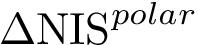 , see Eq. (eq-nispoldiff): variation of , to account for conformational changes upon binding,
, see Eq. (eq-nispoldiff): variation of , to account for conformational changes upon binding, 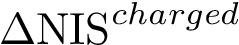 , see Eq. (eq-nischardiff): variation of , to account for conformational changes upon binding,
, see Eq. (eq-nischardiff): variation of , to account for conformational changes upon binding,  , see Eq. (eq-solvation_eisenberg): To challenge amino-acid terms with their atomic counterparts and see which ones are best suited to perform affinity predictions, we also included the atomic solvation energy from Eisenberg et al [83] , describing the free energies of transfer from 1-octanol to water per surface unit (
, see Eq. (eq-solvation_eisenberg): To challenge amino-acid terms with their atomic counterparts and see which ones are best suited to perform affinity predictions, we also included the atomic solvation energy from Eisenberg et al [83] , describing the free energies of transfer from 1-octanol to water per surface unit ( 
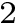 ). The corresponding variable, , is a weighted sum of atomic solvent accessible surface areas (Eq. (eq-solvation_eisenberg)), and may be seen as the atomic-scale counterparts of and .
). The corresponding variable, , is a weighted sum of atomic solvent accessible surface areas (Eq. (eq-solvation_eisenberg)), and may be seen as the atomic-scale counterparts of and . Parameters used to estimate binding affinities.
There are three groups of parameters (see the next equations for their definition):
, , , , , and . , , and . . The acronyms read as follows (see text for details):
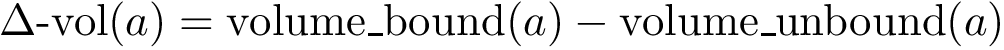
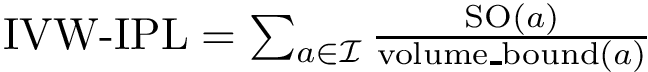
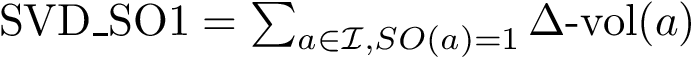
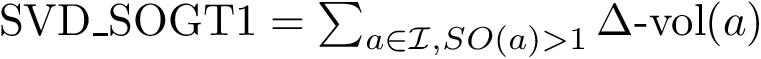
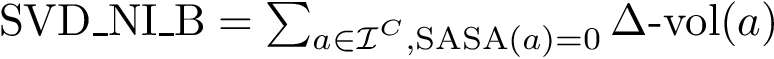
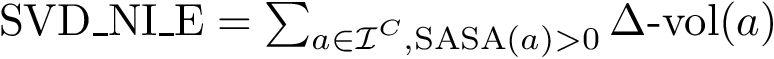
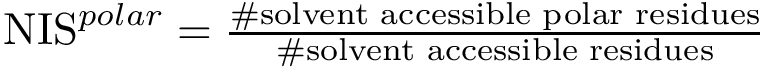
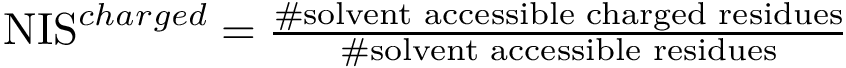
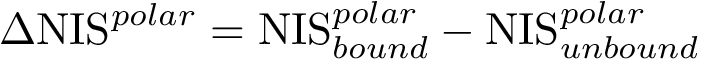

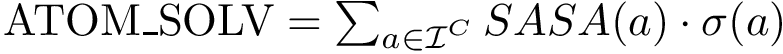
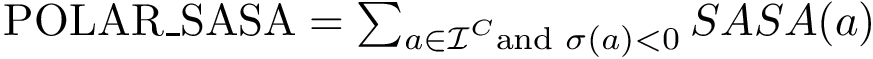
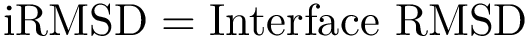
For variables describing a change between the bound and unbound forms of the partners e.g. or , it is necessary to match the atoms of the bound partners to those of the unbound partners. For this, the amino-acid sequences of the partners are extracted from the PDB files and aligned. Then, the atoms of every pair of corresponding residues are matched using their name.
Since missing residues and missing atoms are common in crystal structures, the proportion of matched atoms is computed as to investigate potential wrong matchings, and discard entries for which the alignment is not good enough.
In this section we explain how to compute the variables presented in section Associated variables .
This step consists of two sub-steps:
 : Variables describing geometrical and physico-chemical are computed, using various constructions provided in the SBL. Two settings are supported:
: Variables describing geometrical and physico-chemical are computed, using various constructions provided in the SBL. Two settings are supported:  : Information from the individual runs is assembled. The result may be seen as a matrix of complexes, each defined by its variables.
: Information from the individual runs is assembled. The result may be seen as a matrix of complexes, each defined by its variables.
The input of the pre-processing step consists in:
An XML file complying with the following format:
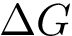 (tag <dG>) (tag <I-RMSD>)
(tag <dG>) (tag <I-RMSD>) Step 1.
The first step of the workflow is performed by the script which is called as follows:
sbl-bap-step-1-run-applications.py -i data/example_data.xml -s data/eisenberg_solvation_parameters.txt -p data/structures -n 1 -d results
are:(-i, –ids-and-chains-file-path) string: input XML file containing the PDB ids and chains of the complexes and unbound partners (required)(-p, –pdb-dir-path) string: sdirectory containing the PDB files to be analyzed (required)(-n, –nb-process) int: number of processes on which to dispatch the execution(-b, –bound-only) bool(=False): toggle computations on the bound structures (complexes) only i.e. not on the unbound partners(-c, –contacts-area) bool(=False): toggle the computation of the area of the Voronoi facets between atoms and residues (uses  )
)(-x, –executables-path) string(=$SBL_BIN_DIR): path to the directory containing the executables.(-v, –verbose) bool(= False): toggle verbose output
| File Name | Description |
| Plain text annotations file | Eisenberg Solvation Parameters |
| XML file | PDB ids and chains of complexes and unbound partners |
| PDB file | Example PDB file in the input directory of option -p |
Step 2.
The second step of the workflow is performed by the script which is called as follows:
sbl-bap-step-2-compile-molecular-data.py -f data/example_data.xml -d results -o results/affinity_dataset.xml
are:(-f, sab-file-path) string: path to the XML file containing the affinity (and optionally ) data for the entries (required)(-o, –output-file-name) string: name of the output file(-b, –bound-only): bool(=False): if set, the program does not try to fetch data about the unbound partners (to be used in conjunction with the -b option from step 1)(-d, –data-directory) string: path to the directory containing the data (defaults to the current directory)(-v, –verbose) bool(= False): toggle verbose output
Step 1.
Practically, this first step consists in running  (through SBL::PDB_complex_to_unbound_partners_matchings::PDB_complex_to_unbound_partners_matcher ), and on various entries of the dataset. For each entry, a directory named after the following scheme is created in the current working directory: <PDB_ID>_<chains_A>_<chains_B>
(through SBL::PDB_complex_to_unbound_partners_matchings::PDB_complex_to_unbound_partners_matcher ), and on various entries of the dataset. For each entry, a directory named after the following scheme is created in the current working directory: <PDB_ID>_<chains_A>_<chains_B>
Each directory is structured as follows:
1A2K_AB_C |-- 1OUN_A_A_atom_matchings.txt |-- 1OUN_A_A_residue_matchings.txt |-- 1OUN_AB | |-- sbl-vorlume_eisenberg_solvation_parameters__log.txt | |-- sbl-vorlume_eisenberg_solvation_parameters__surface_volumes.xml |-- 1OUN_B_A_atom_matchings.txt |-- 1OUN_B_A_residue_matchings.txt |-- 1QG4_A | |-- sbl-vorlume_eisenberg_solvation_parameters__log.txt | |-- sbl-vorlume_eisenberg_solvation_parameters__surface_volumes.xml |-- 1QG4_C_A_atom_matchings.txt |-- 1QG4_C_A_residue_matchings.txt |-- sbl-vorlume_eisenberg_solvation_parameters__log.txt |-- sbl-vorlume_eisenberg_solvation_parameters__surface_volumes.xml |-- sbl-vorshell-bp-ABW-atomic__AB_ball_shelling_forest.dot |-- sbl-vorshell-bp-ABW-atomic__AB_ball_shelling_forest.xml |-- sbl-vorshell-bp-ABW-atomic__AB_packing.xml |-- sbl-vorshell-bp-ABW-atomic__BA_ball_shelling_forest.dot |-- sbl-vorshell-bp-ABW-atomic__BA_ball_shelling_forest.xml |-- sbl-vorshell-bp-ABW-atomic__BA_packing.xml |-- sbl-vorshell-bp-ABW-atomic__interface_AB.pdb |-- sbl-vorshell-bp-ABW-atomic__interface_BA.pdb |-- sbl-vorshell-bp-ABW-atomic__log.txt |-- sbl-vorshell-bp-ABW-atomic__surface_volumes.xml
This example is for entry 1A2K with partners AB and C. All *_matchings.txt files contain the matchings between atoms or residues of the bound and unbound structures. Directories 1OUN_AB and 1QG4_A are for the unbound partners. All sbl-* files contain the results of the SBL executables. If verbose output is toggled (-v flag), various information will be output on the standard and error output.
Step 2.
| Preview | File Name | Description |
| XML archive | Compiled results of runs of step 1 |
In this section, we explain how to select a binding affinity model maximizing a performance criterion, using the script  . Two points worth noticing are:
. Two points worth noticing are:
In the sequel, we explain how to predict 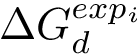 of complexes from a dataset
of complexes from a dataset 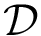 , using the methodology summarized on Fig fig-bap-workflow . Estimation is performed on a per complex basis, from which performances at the whole dataset level are derived.
, using the methodology summarized on Fig fig-bap-workflow . Estimation is performed on a per complex basis, from which performances at the whole dataset level are derived.
Our predictions rely on three related concepts defined precisely hereafter (see also Fig fig-bap-workflow):
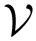 , : the machinery returning one binding affinity estimate
, : the machinery returning one binding affinity estimate 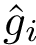 per complex from , using
per complex from , using 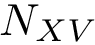 repetitions of the
repetitions of the 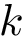 -fold cross validation.
-fold cross validation. Templates.
Denote the pool of variables. Let a template be a set of variables, i.e. a subset of . To define parsimonious templates from the set , we generate subsets of involving up to at most 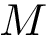 variables. This defines a pool of templates
variables. This defines a pool of templates 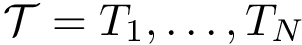 .
.
Cross-validation.
In the following a model is associated to both a template 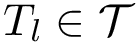 and a dataset . More precisely, a model refers to a regression model, i.e. the variables of the template plus the associated coefficients.
and a dataset . More precisely, a model refers to a regression model, i.e. the variables of the template plus the associated coefficients.
Practically, models are defined during -fold cross-validation, and a number of of repetitions (see Fig fig-bap-workflow). Consider one repetition, which thus consists of splitting at random into subsets called folds. For one fold, a regression model associated with 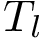 is trained on (k-1)/k of the dataset , and predictions are run on the remaining 1/k of complexes. Processing the folds yields one repetition of the cross validation procedure, resulting in one prediction
is trained on (k-1)/k of the dataset , and predictions are run on the remaining 1/k of complexes. Processing the folds yields one repetition of the cross validation procedure, resulting in one prediction 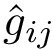 for the of each complex. The set of all predictions in one repeat, say the
for the of each complex. The set of all predictions in one repeat, say the 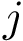 th one, is denoted
th one, is denoted
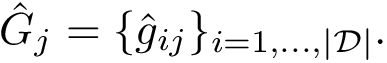
Note again that these predictions stem from regression models associated with , namely one per fold.
Statistics per template.
Considering one cross-validation repetition , we define the correlation 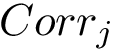 as the correlation between the experimental values
as the correlation between the experimental values 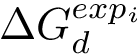 and the predictions
and the predictions 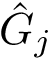 . An overall assessment of the template using the repetitions is obtained by the following median of correlations
. An overall assessment of the template using the repetitions is obtained by the following median of correlations
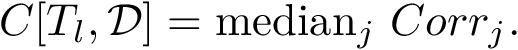
For a complex 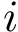 , we define the binding affinity prediction
, we define the binding affinity prediction 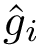 as the median across repetitions i.e.
as the median across repetitions i.e.
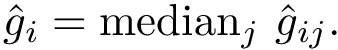
The median prediction error is defined by
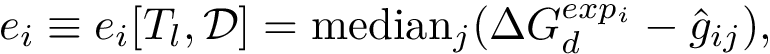
and the median absolute prediction error by:
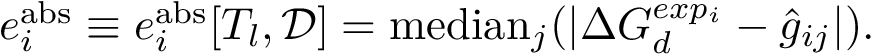
Using this latter value, we define the prediction ratio 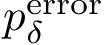 as the percentage of cases such that the dissociation free energy is off by a specified amount
as the percentage of cases such that the dissociation free energy is off by a specified amount 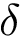 :
:
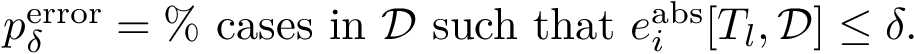
In particular, setting to 1.4, 2.8 and 4.2 kcal/mol in the previous equation yields cases whose 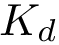 is approximated within one, two and three orders of magnitude respectively.
is approximated within one, two and three orders of magnitude respectively.
Finally, a permutation test yields a p-value for each predictive model [144] .
In a nutshell, the rationale consists of generating randomized datasets by shuffling their values. Then, one computes the performance criterion for each such dataset, from which the p-value is inferred (see Algorithm~alg-p-val).
Model selection.
Define the best predictive model as the one maximizing performance criterion. We wish to single out the best predictive models, i.e. those that cannot be statistically distinguished from the best predictive model. More precisely, consider a pool of predictive models  , out of which we wish to identify a subset of predictive models whose distribution cannot be distinguished from that of the best predictive model. To this end, let H0 be the null hypothesis stating that two predictive models yield identical performance distributions. We decompose the predictive models as
, out of which we wish to identify a subset of predictive models whose distribution cannot be distinguished from that of the best predictive model. To this end, let H0 be the null hypothesis stating that two predictive models yield identical performance distributions. We decompose the predictive models as  such that :
such that :
 , , one does not reject H0, and against one predictive model from
, , one does not reject H0, and against one predictive model from 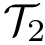 , one rejects H0.
, one rejects H0. The predictive models in are called the specific models for the dataset . The corresponding procedure is based on the Kruskal-Wallis test, see [124] .
The p-value threshold for this test is set to 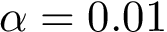 .
.
 |
Running binding affinity predictions for a dataset (Cross-validation) Each template undergoes a number (Statistics) Various statistics are computed to assess the performances yielded by the predictive model associated to each template. (Model selection) Predictive models are compared, and the best ones selected. |
Model generation and selection is performed by the script which is executed as follows :
# Quick example: maximum 2-variables models, 10 repetitions and p-value permutations, # flexible complexes only (irmsd > 1.5 A), run on a single process sbl-bap-step-3-models-analysis.py -f results/affinity_dataset.xml -m 2 -n 1 -r 10 -p 10 -l 1.5 -o flex_quick
# Heavier example: maximum 4-variables models, 100 repetitions and # 1000 p-value permutations, all complexes, run on three # processes. Performs k-nearest neighbors regression with 10 neighbors sbl-bap-step-3-models-analysis.py -f results/affinity_dataset.xml -m 4 -n 3 -r 100 -p 1000 -k 10 -o all_heavier
are:(-f, –data-file-path) string: path to the file resulting from running (required)(-m, –max-order) int: maximum order (i.e. number of variables) to be included in a model (required)(-a, –matching-atoms-filter) float(= 0.8): minimum proportion of atoms matched from a complex structure to its unbound partners structures required for an entry to be included(-n, -nb-process) int(= 1): number of processes on which to dispatch the execution(-d, –nb-folds) int(= 5): number of folds of the cross-validation(-r, –nb-repeats) int(= 1000): number of repetitions of the cross-validation procedure(-p, –nb-pval-permutations) int(= 1000): number of permutations used to compute the p-value of a model(-l, –irmsd-lower-bound) float: minimum allowed for an entry to be included(-s string(= –filter-file-path)): path to the file giving upper and lower bounds on specific variables from the dataset.(-i, –inclusive-bounds) bool(= False): set if the previous bounds should be inclusive instead of strict(-o, –output-file-prefix) string: prefix for the output file(-k, –knn) bool(= False): toggle knn regression instead of linear regression with the given number of neighbors
The XML file specified by option -f should comply with the following format:
The txt file specified by option -s should comply with the following format:
| File Name | Description |
| XML archive file | Compiled data generated from |
This step outputs a minimum of seven files:
Files 1 and 2 are text files with the following structure:
In the first one, the index and the ranks are computed on the cross-validated median absolute error of the model. In the second they are computed on the median cross-validation correlation coefficient.
Files 3 and 4 below contain a matrix of prediction per cross-validation repetition for the best model. Here "best" refers to the model selected by the model selection procedure described above. If multiple models are selected, each one has an associated file. These files are structured as follows:
File 5 and 6: PDF files containing scatter plots of the prediction made by the best predictive models (from files 1 and 2 respectively) for each complex against the experimental affinity value.
is provided. The file contains a scatter plot of the prediction error (Eq. eq-median_error_cplx) made by the best predictive model (correlation-wise) for each complex against its . | Preview | File Name | Description |
| Plain text file | File 1 | |
| Plain text file | File 2 | |
| Plain text file | File 3 | |
| Plain text file | File 4 | |
| PDF file | Scatter plot of the prediction versus experimental affinity for the best model(s) | |
| PDF file | Scatter plot of the prediction versus experimental affinity for the best model(s) | |
| PDF file | Scatter plot of the prediction error versus interface RMSD |
Once a set of variable has been selected during the previous step, it can be used to estimate a model. Recall that a model is a function returning an estimate for binding affinity of a complex. For this, one needs a training set and a regressor, i.e. an object able to use the training set to predict the affinity of a new dataset (for instance, linear least-squares fitting, nearest neighbors, regression trees, ...). An example of the procedure to train such a regressor on the whole Structure Affinity Benchmark and predict its own entries (that is to perform predictions without cross-validation) is given in a demo file (sbl-binding-affinity-model-exploitation-example.py).
We now describe two algorithms used by this package. The first simply computes an approximate (upper bound on the) permutation p-value for a given statistic [144] . The pseudo-code is given on Algorithm alg-p-val .
The second algorithm is used to divide a pool of models into two groups, namely , as discussed in the section Using the programs to select affinity prediction models . The corresponding procedure is based on the Kruskal-Wallis test, and can be found in [124] .

We describe successively the 2 steps of section Using the programs to pre-process structural data .
Step 1.
The script runs applications from the SBL on the bound and unbound partners.
The matching between atoms (section Matchings for atoms) is done using the python module SBL::PDB_complex_to_unbound_partners_matchings which is a wrapper around the executable . This executable performs the matching using the class SBL::CSB::T_Alignment_engine_sequences_seqan, from the package Alignment_engines, which itself uses the Seqan library to perform the sequence alignment, and subsequently match residue and atoms based on the alignment found.
In order to compute shelling orders and packing properties, the executables and are also run on the complex, and is run on the unbound partners.
Step 2.
The script compiles the previous results and computes the variables listed in section Associated variables for each entry of the dataset.
Data structures to store the properties of entities such as atoms, residues, chains and files are grouped in the python module SBL::Protein_complex_analysis_molecular_data_structures . To fill these data structures it also provides a class used to traverse the directory structure and parse the files resulting from step 1.
The machinery used to build, evaluate and rank various models using the previously computed variables can be found in the python module SBL::Combinations_of_variables_model_evaluation::Combinations_of_variables_model_evaluation .
Once a set of variables has been selected, model fitting on a training set and prediction of a new dataset can be done using any software or language. An example script is given using python and scikit-learn in file
The seqan library [73] (BSD, Seqan) is necessary when computing the variables which encode the variations between bound and unbound partners.