|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
![]()
Authors: F. Cazals and T. Dreyfus and S. Marillet and R. Tetley
Define an alignment engine as an alignment algorithms producing suitable statistics. This package unifies the two main ways alignments are done in bioinformatics, namely sequence alignments and structural alignments:
sequence alignments aim to match units (amino acids or nucleotids) of multiple sequences only regarding their positions in the sequences and some similarity between these units. Typically, a substitution score matrix determines the penalties for matched units that are not identical, or for creating or prolonging gaps (i.e, sequences of units that do not match any unit in the other sequences).
Defining a common framework for both allows factoring out common bits, in particular statistics on the aligned sequences. Note also that this package does not provide any algorithm, per se, as sequence and structural alignments are borrowed from external libraries.
Alignment engines are designed so that it is easy to wrap an existing sequence or structural alignment algorithm into the engines. In order to do so, we first establish the terminology used in the package, then explain the design choices.
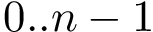 convention. If each unit is also equipped with cartesian coordinates, it is geometrically embedded and the sequence may also be called a structure.
convention. If each unit is also equipped with cartesian coordinates, it is geometrically embedded and the sequence may also be called a structure.
The alignment engines are designed so has to be independent from the aligners, meaning that aligners are template parameters of the engines. For this reason, the main difference between structural and sequence alignment engines are the analysis : structural sequence alignment engines provide geometric analysis in addition to all the analysis provided by the sequence alignment engines.
For this reason, structural alignment engines are seen as specializations of sequence alignment engines. Thus, there are two main classes :
SBL::CSB::T_Alignment_engine< SequenceOrStructure , AlignerAlgorithm , FT > : the base class for sequence alignments. The parameter SequenceOrStructure is the representation of a SOS, AlignerAlgorithm is the aligner itself, and FT is the number type used for rendering the analysis.
SBL::CSB::T_Alignment_engine_structures< StructureType , AlignerAlgorithm , FT , MolecularDistance > : the base class for structural alignments. The parameter StructureType is the representation of a structure, AlignerAlgorithm is the structural aligner itself, FT is the number type used for rendering the analysis, and MolecularDistance is a distance between two structures for comparing the output alignments.
The different template parameters have a number of requirements :
SequenceOrStructure : defines the base types used in the engine (Alignment_unit, Alignment_unit_name for the name of the unit, and Alignment_unit_rep for the index of the unit), and provides an array operator for accessing to any unit of the SOS at a given index. The type Alignment_unit must provide the method get_name() returning the name of the unit.
StructureType : in addition to the requirements for SequenceOrStructure , the type Alignment_unit has to also provide the methods x(), y() and z() returning the cartesian coordinates of the unit.
AlignerAlgorithm : a simple functor taking as argument references to the two SOS to align, and returning the score of the algorithm together with the alignment.
FT : there is no particular requirement, and the default type used is double.
MolecularDistance : see the package Molecular_distances for a complete description of this parameter. It is set by default to the class SBL::CSB::T_Least_RMSD_cartesian.
Both engines are generic and can handle a variety of aligners. For each alignment engine, a specialization with a given aligner is provided:
SBL::CSB::T_Alignment_engine_sequences_seqan< SequenceType , SeqanSequenceConverter , FreeEndsAlignment , ScoreType , SeqanUnitType , SeqanCustomMatrix , SeqanAlgorithm > : specialization of the sequence alignment engine with aligners from the Seqan library. The parameter SequenceType has the exact same requirements as the previous parameter SequenceOrStructure . The parameter SeqanSequenceConverter is a functor converting the name of a unit in SequenceType to the name of a unit in Seqan (by default, it does nothing). All other parameters are specific to Seqan, and we recommend the user to visit the documentation of the Seqan library.
SBL::CSB::T_Alignment_engine_structures_apurva< StructureType , FT , MolecularDistance > : specialization of the structural alignment engine with aligners from Apurva [11] . All parameters have been already described previously.
There are three sets of functionality :
the generic alignment analysis : all engines provide at least the score of the aligner (depending on the aligner algorithm), the percentage of similarity (that is the score obtained by summing the substitution score of paired entities) and identity (that is the percentage of similarity for the identity substitution matrix) of an alignment given an input substitution matrix, and the possibility to view the alignment using Graphviz .
the structural alignment analysis : structural alignment engines provide in addition the Distance Difference Matrix (DDM), the dRMSD computed from the DDM, and the molecular distance between the two aligned structures.
the modules : structural and sequence alignment engines are embedded in two modules for a simple use within the Module framework of the SBL : SBL::Modules::T_Alignment_sequences_module and SBL::Modules::T_Alignment_structures_module
In addition of these functionality, the program  provides the following functionality:
provides the following functionality:
1-to-1 matching : given two atomic models of two polypeptide chains, the program matches the residues with a sequence alignment, then matches the atoms of matched residues; results are stored in two files, one with the residue ids of the matched residues, the other one with the atomic serial numbers of the matched atoms;
We consider a set of sequences and possibly associated structures; 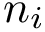 stands for the number of residues in the structure, and
stands for the number of residues in the structure, and 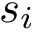 the length of the associated primary sequence.
the length of the associated primary sequence.
The class SBL::CSB::T_Multiple_sequence_alignment_DS to store MSA provided FASTA files.
In working with MSA, one must handle - symbols which account for unaligned units. This is done using two mechanisms:
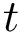 dashes in the corresponding MSA column. The number of valid positions is denoted
dashes in the corresponding MSA column. The number of valid positions is denoted 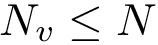 , with
, with 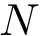 the length (number of columns) of the MSA.
the length (number of columns) of the MSA.
If 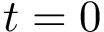 , a column in the MSA is valid iff no sequence contains - at this position.
, a column in the MSA is valid iff no sequence contains - at this position.
In using MSA, three types of indices are of interest:
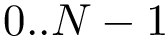 ,
, 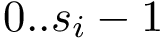
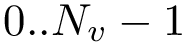 .
.Consider the following types:
Using them, the following data structures are provided to navigate between alignment indices, sequence indices, and valid alignment indices:
Practically, one calls the function m_msa->get_valid_length() to get the number 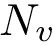 of valid positions. Then, with
of valid positions. Then, with 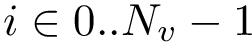 , the sequence index is obtained via
, the sequence index is obtained via
An important case is that where the sequences of the MSA are accompanied by structures.
The class SBL::CSB::T_Chains_residues_contiguous_indexer is instantiated from a set of structures – e.g. polypeptide chains of the package Protein_representation. For each sequence/structure, it contains two pieces of information.
Structural information. Assume the i-th chain has gaps–some residues have not been reported. In that case, 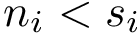 . The contiguous index of a residue is an integer in the range
. The contiguous index of a residue is an integer in the range 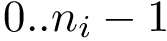 .
.
For each sequence/structure, the class SBL::CSB::T_Chains_residues_contiguous_indexer records a map from 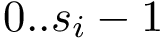 to : it maps a resid (minus one to be precise) to a contiguous index in
to : it maps a resid (minus one to be precise) to a contiguous index in 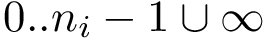 . This map can be queried to know whether a particular residue is present in the structure.
. This map can be queried to know whether a particular residue is present in the structure.
Sequence information. A sequence can be retrieved from the seqres field of the structure file. Or it can be built while parsing the atoms of the structure. In this latter case, if 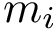 is the largest residue sequence number in the i-th structure, the corresponding sequence has length . Absent residues are denoted with the letter X in the sequence.
is the largest residue sequence number in the i-th structure, the corresponding sequence has length . Absent residues are denoted with the letter X in the sequence.
Def. def-valid-msa focuses solely on sequences in the MSA. We enrich it to account for the presence of residues in structures:
The class SBL::CSB::T_Multiple_sequence_alignment_DS_filtered specializes SBL::CSB::T_Multiple_sequence_alignment_DS. In particular, the function compute_nb_missing_residue_per_position() which is used to identify the valid position, incorporates the call to a functor providing the is_missing() function:
See the section below for a toy example using such a functor.
A non trivial example is provided in the package Spectrus-package, where valid positions in the MSA are invalidated if one (or several) structures miss a particular residue, using an instance of the class SBL::CSB::T_Chains_residues_contiguous_indexer .
The following examples show how to use the two specializations of the alignment engines with Seqan and Apurva, and how to use the two provided modules.
This example loads an input set of polypeptidic chains from two PDB files and align their sequences. Then it dumps the aligner score from Seqan, together with the identity and similarity percentages. Note that a fifth argument is required corresponding to the occupancy policy, i.e how atoms with an occupancy lower than 1 in the PDB file should be treated.
See the reference manual of SBL::CSB::T_Alignment_sequence for the definition of the Sequence data structure.
This example loads an input set of polypeptidic chains from two PDB files and align their structures. The coordinates of the residues are the coordinates of the carbon 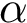 of the residue. Then it dumps the aligner score from Apurva, the identity and similarity percentages, and the dRMSD and the lRMSD of the output alignment. Note that a fifth argument is required corresponding to the occupancy policy, i.e how atoms with an occupancy lower than 1 in the PDB file should be treated.
of the residue. Then it dumps the aligner score from Apurva, the identity and similarity percentages, and the dRMSD and the lRMSD of the output alignment. Note that a fifth argument is required corresponding to the occupancy policy, i.e how atoms with an occupancy lower than 1 in the PDB file should be treated.
Note that Apurva can filter the alignment with Secondary Structure Elements (SSE), if these are provided. The Sequence data structure is designed so that Apurva uses the SSE information in the PDB file to filter the alignment.
This example is similar to the example in section Sequence alignments with Seqan, except that it uses a module to perform the work. In particular, the alignment and the analysis are done within the module. It also offers the possibility to report the alignment into a Graphviz file.
This example is similar to the example in section Structural alignments with Apurva, except that it uses a module to perform the work. In particular, the alignment and the analysis are done within the module. It also offers the possibility to report the alignment into a Graphviz file.
We provide a toy example to compare the data structures SBL::CSB::T_Multiple_sequence_alignment_DS and SBL::CSB::T_Multiple_sequence_alignment_DS_filtered .
Toy example. To describe the processing done in sbl-test-msa-ds.cpp (see snippet below), consider the following alignment between two chains of length 7:
> cat ../../../data/fictitious-2prots.fasta >A AAA-AAA >B A-AAAAA
Also consider the following example filter, which retains even columns of the MSA
Upon building instances of the classes MSA_one_letter_code and MSA_one_letter_code_filtered on our toy alignment with two sequences, we get:
=== MSA Chain and size:A 6 AAA-AAA Chain and size:B 6 A-AAAAA File chain is A; 7 indices in the table m_alignment_index_to_chain_index 0 1 2 4294967295 3 4 5 File chain is B; 7 indices in the table m_alignment_index_to_chain_index 0 4294967295 1 2 3 4 5 File chain is A; 5 indices in the table m_valid_alignment_idx_to_chain_idx 0 2 3 4 5 File chain is B; 5 indices in the table m_valid_alignment_idx_to_chain_idx 0 1 3 4 5 === MSA-filtered Chain and size:A 6 AAA-AAA Chain and size:B 6 A-AAAAA File chain is A; 7 indices in the table m_alignment_index_to_chain_index 0 1 2 4294967295 3 4 5 File chain is B; 7 indices in the table m_alignment_index_to_chain_index 0 4294967295 1 2 3 4 5 File chain is A; 3 indices in the table m_valid_alignment_idx_to_chain_idx 3 4 5 File chain is B; 3 indices in the table m_valid_alignment_idx_to_chain_idx 3 4 5
Note in particular that if one filters out - and position for which sequences indices are less than 2, sequences indices 3, 4 and 5 only remain valid. (NB: sequence indices are sequence specific and run from 0 to 5 for each of our two toy sequences.)
See the snippet