|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
Tutorial delineating the structure of the documentation of the library.
In the sequel, we outline the structure of the user and reference manuals for each main part of the library (Applications, Models and Core).
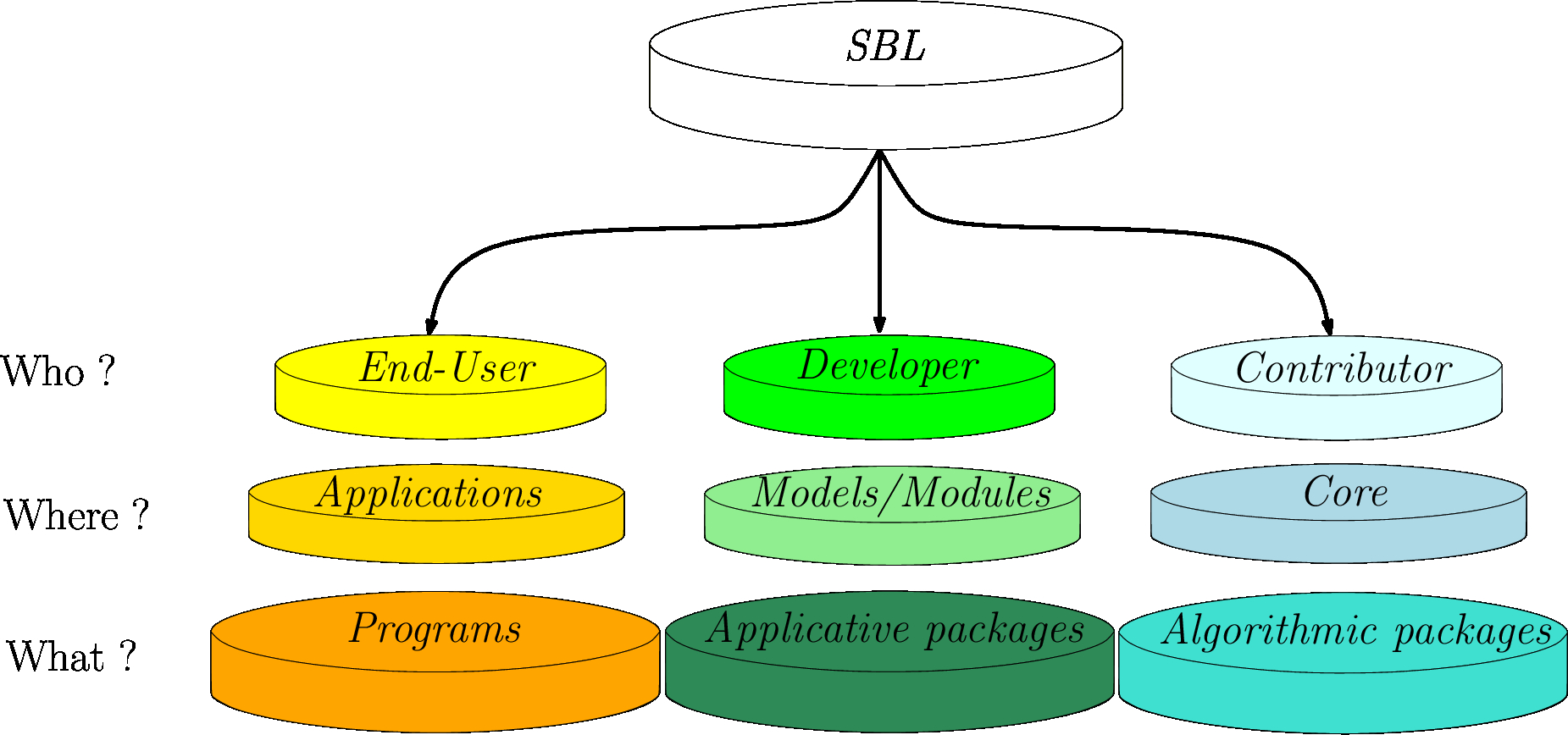 |
| Overview of the SBL: main focus areas for end-users, developers, and contributors. |
The documentation of the applications is accessible from the home page, in the section Introduction, or on the side bar by developing the Structural Bioinformatics Library item, then the Packages item, then by clicking on the Applications item. On the Applications resulting page, different categories are displayed, together with the programs available in these applications.
A given application comes with a short description and a link to its reference and user manuals. The former discusses the problems solved by the application, and the basic programs used. (NB: since an application comes with generic C++ code, it may be instantiated to generate new programs solving the same problem, but with different data.) The latter discusses in detail the classes specifically used for the application.
In the following, we present
An application package – see Applications, defines a set of utilities i.e. computer programs solving related problems, or solving the same problems with different algorithms. We illustrate this with two examples:
The user manual is articulated around the following sections:
Goal: a light and user friendly presentation of the functionalities provided by the application.
Using the Application: example calls of the programs, with example input files and options. The input and output files are accessible from the <install_directory>/share/doc/SBL/demos.
Pre-requisites: a formal presentation of the objects manipulated, either as input or output.
Algorithms and Methods: a concise description of the algorithms underlying the application, and bibliographical pointers if appropriate.
Programmers's Workflow: a concise description of the C++ classes (and their template parameters) used in the implementation of the application. This is meant to ease the development of twin applications, using different types (in C++ terminology: different C++ models matching a given C++ concept), or undertaking similar tasks.
Programmer's workflows are automatically generated by the programs and are represented using UML-like diagrams. An example workflow for computing the volume and surface area of a molecule is presented on Fig. fig-example-workflow. The gray rhombus represents the start and the end of the application:
the out-going edges from the start indicates the first steps (without particular order) that will be processed. When some steps are optional, the options to set for processing these optional steps are indicated with the start rhombus.
The white ellipses indicate the modules (see Modules), whose main features are:
A module is a step transforming an input into an output.
A module always has a single input. If several inputs are required, one needs to use binary logical operators (and , or), that are represented by white triangles in the workflow: a logical operator has always two in-going edges and one out-going edge.
When modules have options, these options are listed under the name of the step. Optional modules are indicated by a dashed white ellipse.
In C++, a concept is a set of requirements that a type must fulfill to be usable in a given algorithm, and a model matching a concept is a type fulfilling those requirements, see Boost concepts check library.
Within the SBL, this mechanism is used in Models to bridge the gap between biophysical data and algorithms. That is, the models can be seen as sets of requirements imposed by generic algorithms. The SBL models are grouped within by concept within packages. The concepts are always related to biophysics, as illustrated with the two following examples:
The user manual is articulated around the following sections:
Introduction: a definition of the SBL concept with the notations used in the package.
Using existing SBL models in existing Applications: a section for end-users, illustrating the use of programs involving a model of the SBL concept discussed.
Using existing SBL models for new Applications: a section for developers, listing the main C++ models provided for the SBL concept discussed, and discussing the integration of such models within an application.
Developing new SBL models: a section for developers, listing the requirements of the SBL concept discussed–these requirements must be implemented by any model following this SBL concept.
Examples: a list of the examples referenced in this user manual
A package in Core solves a specific problem by providing algorithms and / or data structures. The Core packages are divided in several groups depending on the problem they solve – see main page of Core . CADS , GT and CSB . Each part has its own namespace. While a major part of the classes in a package are in the corresponding namespace, it is not necessarily the case for all the classes of the package. This is illustrated with the two following examples:
 -complex of a molecular model, where the particles of this model are represented by 3D balls. Since it provides a data structure in a biophysical context, i t is a package of CSB . While the main classes are embedded in the namespace CSB, classes and methods for serializing and visualizing the -complex are embedded in the namespace IO . Furthermore, there is an additional class corresponding to a module – i.e a black box facilitating the computation, analysis and serialization of the -complex, that is embedded in the namespace Modules .
-complex of a molecular model, where the particles of this model are represented by 3D balls. Since it provides a data structure in a biophysical context, i t is a package of CSB . While the main classes are embedded in the namespace CSB, classes and methods for serializing and visualizing the -complex are embedded in the namespace IO . Furthermore, there is an additional class corresponding to a module – i.e a black box facilitating the computation, analysis and serialization of the -complex, that is embedded in the namespace Modules .The user manual is articulated around the following sections:
Introduction: definition of the problem solved by the package together with the algorithms and data structures used for solving it.
Implementation: a presentation of the main classes of the package.
Examples: a list of the examples referenced in this user manual
The reference manual of a package lists the classes specific to that package. The composition differs depending on the main part of the package:
Applications : one need to document (i) the workflow class where is designed the workflow of the application as a graph connecting modules, (ii) the traits class defining the types used in the workflow class, and (iii) the specific modules used in the corresponding application.
Models : one needs to document all the models of the concept of the corresponding package.
While the reference manual lists all classes of the packages with no distinction, the guard page of each package lists the most important classes of that package. A user should first check the main classes before inspecting all other classes.