|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
![]()
Authors: F. Cazals and T. Dreyfus and R. Tetley
Structural alignments.
Given two structures, a structural alignment is a matching between structural motifs within these structures. The problem has many variants, depending on (i) the representation used, (ii) the score optimized, and (iii) the optimization algorithm used.
As typical examples, one may cite:
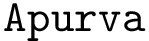 is a structural aligner based on contact maps, favoring long and flexible alignments [11] – see Apurva.
is a structural aligner based on contact maps, favoring long and flexible alignments [11] – see Apurva.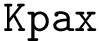 is a structural aligner based on a geometric representation of the backbone, favoring a geometric measure known as the G-score [151] .
is a structural aligner based on a geometric representation of the backbone, favoring a geometric measure known as the G-score [151] .Iterative alignments. Iterative alignments [24] , [151] consists in iteratively finding the alignment for fixed positions of the conformations–via dynamic programming, and finding optimal superimposition given the alignment–the classical rigid superimposition problem. The process is typically iterated until a fixed point or a cycle is reached. Such alignments are especially interesting when seeded with our motifs, for two reasons.
Performing a structural alignment has two components:
Each step is easy given the result of the other one:
The complexity actually comes from solving these two steps at once. To do so, a classical strategy consists in interleaving the DP two operations just discussed.
This is used in the fuzzy alignment from [24] , and also for [151] .
Alignments and statistics. The companion package Alignment_engines provides tools to compute statistics on various alignments.
The Iterative_alignment package provides a number of novel classes to ease the instantiation of novel aligners – see e.g. the package Kpax .
The classes from this package and those from the package Alignment_engines are used to implement the algorithm, see Kpax .