|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
|
Structural Bioinformatics Library
Template C++ / Python API for developping structural bioinformatics applications.
|
![]()
Authors: F. Cazals and T. Dreyfus
In the SBL, a molecule of N atoms may be considered following two aspects :
combinatorialy, atoms of a molecule are linked together forming a graph where atoms are vertices and bonds between atoms are edges of this graph;
geometrically, atoms of a molecule may be considered as 3D points so that the molecule itself may be considered as a D-dimensional point in a space of dimension 3N. graph;
Such a D-dimensional point is called a conformation, and this package provides data structures for representing conformations. More precisely, it provides a unique Traits class listing types and static methods, that is definable for any data structure that could represent a the conformation. In this way, any data structure that could fit the concept of a conformation can be used by simply implementing this unique Traits class for this target data structure. Technically, this feature is called Partial Template Specialization .
When using applications of the SBL dealing with conformations, a critical matter is the way conformations are stored, and how to load them.
The conformation of a molecule involving 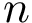 particles can be described by
particles can be described by 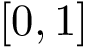 Cartesian coordinates. Those coordinates can be stored in a text file following the D-dimensional point format : the number of coordinates, called the dimension, is followed by all the cartesian coordinates. Note that the format does not depend on line breaks, tabulations or spaces, so that both following formats are valid – the latter being more user friendly / readable:
Cartesian coordinates. Those coordinates can be stored in a text file following the D-dimensional point format : the number of coordinates, called the dimension, is followed by all the cartesian coordinates. Note that the format does not depend on line breaks, tabulations or spaces, so that both following formats are valid – the latter being more user friendly / readable:
6 0 0 0 1 1 1
or
6 0 0 0 1 1 1
Programs of the SBL requiring molecular conformations are dealing with flexible molecules, posing the problem of loading multiples conformations. To represent such conformations, we decouple the information information as follows:
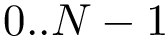 , uniquely identifies a conformation.
, uniquely identifies a conformation.This design has several advantages:
For example, consider a tree connecting the conformations. Each node of the tree contains an index referencing a conformation. The coordinates of that conformation are recorded separately in some container (list, vector, matrix), and are accessed from that container.
Thus, programs of SBL dealing with an ensemble of conformations allow to load conformations in two ways :
In the second case, it is possible to control which atoms are loaded by switching tags using command-line options (in particular : –water for water molecules, hydrogens for hydrogens, hetatoms for hetero-atoms). It is also possible to save the loaded conformations from the PDB files into a D-dimensional point plain text file through the option –save-ensembles.
The conformations used may not have any molecular semantic. For example, the Landscape_explorer can be used to explore polynomial functions rather than the potential energy of a given molecule. In such a case, conformations can be used without any biological meaning, and a natural choice is to represent a conformation with a D-dimensional points. Such programs usually offer a restricted set of options, in particular it is only possible to load conformations from a plain text file listing D-dimensional points.
A model of a conformation can virtually be any type, provided that the class SBL::Models::T_Conformation_traits< Conformation > is defined for this type. A simple STL vector of float can be a conformation by include the file SBL/Models/Conformation_traits_vector.hpp that partially defines the class SBL::Models::T_Conformation_traits< Conformation > for STL vectors. Two important models are implemented :
STL vectors, in the file SBL/Models/Conformation_traits_vector.hpp, for a simple representation
In some circumstances, a more complete data structure is required :
when the conformation is elevated with a height – which may e.g. represent a potential energy, this height needs to be attached to the conformation; the filesSBL/Models/Conformation_traits_with_height.hpp and SBL/Models/Conformation_traits_with_implicit_height.hpp partially defines the data structures SBL::Models::T_Conformation_with_height and SBL::Models::T_Conformation_with_implicit_height and traits class over these data structures.
Note that in the cases above, the data structures are minimalistic and can be easy replaced by any other data structures, provided the right traits class. For exemple, if one wants to use the type std::list<double> as a conformation type (not recommanded due to the random access time), one has to define the class SBL::Models::T_Conformation_traits<std::list<double>> with the types and static methods :
There are two particular cases where the conformation needs to be enriched :
Some parts of the SBL also requires I/O operations over the conformations, and the operators << and >> needs tobe defined for printing / loading the conformations. This is the case when loading the conformations using the package MolecularGeometryLoader. The SBL uses heavily the Boost Serialization library for serializing to / deserializing from XML archives : some modules require to define the the global method serialize(ar, C, flags), where ar is the archive where is serialized / deserialized the conformation, C is the conformation, and flags is a long integer flag for versioning the archive. See Boost Serialization for more details.
- Partial serialization: the DS is serialized, but an index is dumped for each conformation – instead of its full coordinates.
See the package Multiple_archives_serialization for more details.